論文:物質使用障害におけるGABAA受容体サブユニットの発現変化について
原文:Regulation of GABA A Receptor Subunit Expression in Substance Use Disorders PMID: 32580510
投稿:2020年6月22日
Abstract
神経細胞の発火は、神経伝達物質GABA(γ-アミノ酪酸)を媒介とし、その放出によってうまく調整されている。イオンチャネル型GABAA受容体(GABAAR)は、5つの異なるサブユニットで構成されており、脳の領域と細胞タイプによってその発現は異なる。GABAA受容体に対するGABAの作用は、ベンゾジアゼピンやアルコールなど、臨床および薬理学的に重要なさまざまな物質によって変調する。これらの物質への暴露は恒常性を破壊し、GABA作動性神経伝達の可塑性を誘発する。この研究で我々は、病理学的可塑性および適応の観点から、GABAARサブユニットの発現調節を物質使用にフォーカスしながら確認していくことにした。調査の範囲は以下におよぶ。5' 非翻訳領域と3' 非翻訳領域におけるレギュレーション、DNAメチル化における変化、サブユニット発現を調節する初期遺伝子と転写因子、翻訳および翻訳後の変性、その他の受容体変性形態。
神経適応、可塑性という観点から、物質使用およびその離脱中に変異するGABAA発現調節への理解を深めることにより、物質使用障害におけるGABA作動性シグナル伝達の役割について洞察を得、新しい標的療法(novel targeted therapies)の開発に貢献したい。
Keywords: GABA; GABAA receptor; expression; transcription; translation; substance use; substance
use disorder; alcohol; benzodiazepines; plasticity
1. イントロダクション
脳の神経発火パターンは、抑制系神経伝達物質によってパワフルに調節される。
-アミノ酪酸(GABA)。 GABAA受容体(GABAAR)は仲介するリガンド依存性イオンチャネルであり、脳における高速抑制系神経伝達のほとんどを仲介している。GABAAR機能障害は、不安、てんかん、および物質使用傷害などの神経学的および精神医学的疾患に関連する。GABAARは、5つの異なるサブユニットからなるヘテロペンタマーで、脳領域、細胞型、および細胞内ドメインによってその発現が異なる。そして、相同アミノ酸配列によってサブクラスに分類された受容体サブユニットが、少なくとも19ある;α1~6, β1~3, γ1~3, δ, ε, π, θ, そして ρ1~3[1]。受容体サブユニットの組み合わせと機能はさまざまであるが、ほとんどはαふたつ、βふたつ、およびガンマひとつで構成されると言ってよい[2,3]。GABAARサブユニットの発現もまた、mRNAが支配的な初期発達時に、GABAARサブユニットα2, α3, α5, β3の発現とともに暫定的にレギュレーションされる。そして成人するとα1, α4, β2, およびδサブユニットに交代する[4].。 この変化は塩化物の逆転電位の切り替えと同時に発生し、GABAを脱分極から過分極へ切り替える[4,5]。
特にαサブユニットは受容体の局在と機能の重要な決定因子である。α1,β2,γ2構成のGABAARは成人の脳ではもっとも広く発現するものであり、そしてα1サブユニットがもっとも多く発現する[2,4,6].。α1, β1~3, γ2の免疫反応性が脳全体に見られる。α2を含む受容体は小脳、前頭葉、海馬に豊富に存在し、α4を含む受容体は線条体と視床下部に、α5を含む受容体は嗅球と海馬に、α6の免疫反応性は小脳顆粒細胞と蝸牛神経核に比較的限定されている[7、8]。
サブユニット発現は細胞内の局在によっても異なり、主にα1〜3がシナプス部位で多くを占める。α4~6はシナプス外に局在する。シナプスGABAARはまた、介在ニューロンのシナプス前コンパートメントが樹状突起、体細胞、軸索の最初のセグメントに接触する、複数のシナプスサブタイプにも関連している。シナプスおよびシナプス外細胞内ドメインに至るまで、特定のGABAARサブタイプのクラスタリングは、ゲフィリン、コリビスチン、ジストロフィンなどの足場タンパク質をともなうサブユニット固有の相互作用によってレギュレーションされていると考えられている[10–12]。
アゴニストの親和性、ゲーティング、および薬理学的特性の違いが、GABAARサブユニットのコンビネーションを変更することによって繰り返し確認されている[13-16]。サブユニット構成はGABA効力に影響する、例えばα2およびα3を含む受容体はGABA効果が低く、α1,α4,α5だと中程度の効力、α6では高い効力を示す。 GABAARは、臨床的に使用する多くの薬物の作用部位であり、ほとんどは薬物はGABAARにとってポジティブアロステリックモジュレーターとして作用する。そして抑制性のシナプス後電流(inhibitory post-synaptic currents. IPSCs)の減衰を長引かせることにより、段階的抑制を変更する。このことにより、翻って興奮性刺激に反応し神経発火を防ぐこととなる[13、18]。シナプス後GABAARでのIPSCの動態は、次のような生物物理学的特性によって決定される。サブユニットの構成とその細胞膜における分布[18]、受容体機能の因子を決定しIPSC動態を設定する特定のαサブユニット[13、18〜20]。
GABA作動性シグナル伝達は、神経伝達物質合成、小胞の貯蔵、神経伝達物質の放出と再取り込み、シナプス後受容体のクラスタリング、といった変化により細胞レベルで制御される [1]。細胞レベルでのGABA作動性シグナル伝達は、しばしばGABAARの発現変更により変動する。GABAAR遺伝子発現は、一生を通して人の体験、薬物使用、多くの神経病理によってレギュレーションされている[4、21、22]。GABAARサブユニットをエンコードする遺伝子の発現は、転写開始、オルタナティブスプライシング、mRNAの安定性、翻訳、翻訳後修飾、細胞内追跡およびタンパク質分解など複数のレベルで変更され得る[1](図1)。本稿では、GABAAR発現調節について、物質使用とその離脱に焦点を当てながら、正常状態と病的状態の双方からすでに解明されていることについて述べる。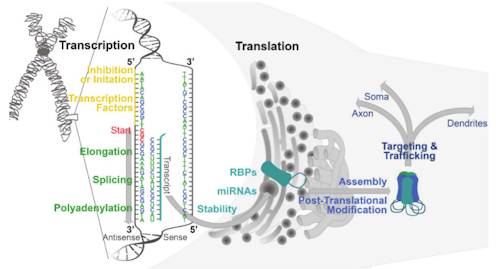
図1.GABAARの発現と機能に影響を与えるレギュレーションの仕組み
サブユニットは転写の開始で始まり、多タンパク質複合体によって制御される。多タンパク質複合体は個々のサブユニットをエンコードする50UTR隣接遺伝子を調節する。転写を調節する転写因子およびその他のタンパク質(黄色)は、50UTRのコンセンサスサイトに結合し開始を促進または抑制する。多くのGABAARサブユニット50UTRには、複数の転写因子のコンセンサスサイトと、複数の転写スタートサイト(赤)が含まれている。スプライシングとポリアデニル化という補足的プロセス(緑色)が、リボソーム(灰色の円)での翻訳のために、転写物を粗い小胞体にシャトルする前に核で発生する。サブユニット転写産物の翻訳は、RNA結合タンパク質(RBP;ターコイズ)およびマイクロRna(miRNA;ターコイズ)によって調節されることが示されている。翻訳されると、サブユニットは四次構造で組み立てられ(青色)、機能的なヘテロ五量体GABAARを形成する。 GABAARはまた、リン酸化やパルミトイル化などの翻訳後修飾(青)による調節対象でもあり、特定の細胞内ドメインに優先的にローカライズされるように調節され、ターゲティングやトラフィッキングも行われることになる。
2. GABAA受容体の発現を調節する基本的なメカニズム:転写、翻訳、その他
物質使用におけるGABAAR発現の動的調節を理解するために、基礎生理学的状態下でのサブユニット発現調節メカニズムを調べることが重要となる。転写、翻訳、そしてGABAAR発現の正確なチューニングに関わるその他多くのレギュレーション要因を調べることにより、多くのことが明らかになってくる。次のセクションでは、転写因子、エピジェネティックレギュレーター、およびGABAARサブユニット発現を調節するRNA結合について要約していく。
GABAARとそのサブユニットの進化は、系統樹と染色体上の遺伝子構成を直接検査することでアクセス可能である[23]。サブユニット遺伝子の多くは、異なる染色体上のβ-α-α-γとβ-α-γクラスターで変性されており、単一の祖β-α-γから進化したと考えられている。合計すると、GABAARサブユニットをコードする4つの遺伝子クラスターが人体で発見され、この組織はおそらく協調遺伝子調節に関与していると考えられている[1,24]。β1、α2、α4、およびγ1(GABRB1; GABRA2; GABRA4; GABRG1)をコードする遺伝子はすべてヒトの染色体4p14-q12にクラスター化されている[25、26]。β1、α2、α4サブユニットはすべて成体ラットの海馬で濃縮されており、クラスターの組織化が領域固有の発現を維持するために必要である可能性を示す[1,6]。染色体5q32.1-q35には、最も一般的なGABAARサブユニットβ2-α1-α6-γ2 [27](GABRB2; GABRA1; GABRA6; GABRG2)のクラスターコーディングが含まれており、サブユニット遺伝子のクラスター化が発現調節の役割を果たすこと、および共通因子が複数のサブユニット遺伝子発現を調節する可能性があることを、やはり示唆している
2.1 転写
mRNAレベルと遺伝子発現のコントロールについて、主たるフェーズとなるのは転写開始(transcription initiation)である。これには、多くの非翻訳領域(UTR)で、遺伝子を取り巻く個別のモチーフを認識する複数の転写因子と、その他DNAにバインドするタンパク質のオーケストレーションが必要である[28]。アクティブなクロマチンにおいて、遺伝子の50末端に隣接する保存されたDNAモチーフは、DNA結合タンパク質の多様なセットと相互作用し(図1)、さまざまな生理学的刺激に応じて変化する。酵母とショウジョウバエの遺伝的アプローチ、および哺乳動物細胞の生化学的試験法を用いて、転写開始に必要なRNAPol II複合体の形成に役立つ、配列特異的活性化因子とアクセサリー因子の大きな集合体が確認されている[28]。転写制御の理解については世界的な進展があるが、GABAARサブユニットをコードする遺伝子を含め、個々の遺伝子の詳細で精巧なコントロールについてはほとんどわかっていない[28]。
シリコ分析実験(silico analysis and experimentation)が、コア要素、可能な近位転写因子、および転写開始部位の予測を含むGABAARサブユニット遺伝子のプロモーターを調べるために使用されている(図2)[29,30]。複数の種からのアラインメント分析により、特異性タンパク質(Sps)の結合部位やcAMP応答要素結合タンパク質(CREB)の結合部位など、GABRA1に隣接する高度に保存された50配列が示された[29] 。Spsは、多くの遺伝子発現を調節することで知られているジンクフィンガー転写因子( zinc-finger transcription factors )である。ニューロン固有の転写因子Sp4は、GABRA1の発現と興奮性神経伝達物質受容体遺伝子を調節することが示されている[31]。 CREBと誘導性cAMPアーリーリプレッサー(ICER)は、α1サブユニットの転写調節因子であると実験で確認されている[32]。CREBは、可塑性のメカニズムに関与している刺激誘発転写因子である[32、33]。CREBは学習と記憶において確固たる役割を持ち[34]、物質使用反応にも関与している[35,36]。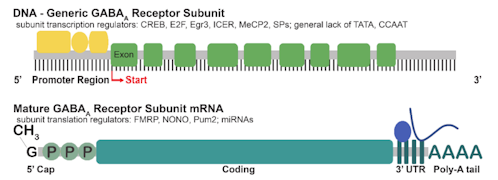
図2. GABAARサブユニット発現に影響を与える転写および翻訳の主要な調節因子
一般に、GABAARサブユニット遺伝子の50 UTRには、転写因子の複数のコンセンサスサイト(黄色)、および多くの場合、複数の転写開始サイト(赤)が含まれている。サブユニットの大部分には、50UTRに標準のTATAおよびCCAATシーケンスが存在しない。多くのレギュレーターがCREBやMeCP2を含む複数のサブユニットの転写に影響を与えることがわかっている。mRNAの成熟には、50キャップと30UTRに隣接するポリAテールも含まれる。成熟したGABAAR転写産物は、核からラフな小胞体のリボソームに輸送され、そこでは、翻訳はFMRP、NONO、Pum2などのRNA結合タンパク質(青い楕円)や、microRNA(miRNA;青い線)の影響を受ける。
GABRA2遺伝子は、複数のスタートサイト、3つのプロモーター領域を持ち、エクソン1A、1B、および1Cの選択的スプライシングから生じる6つのmRNAアイソフォームを生成することがわかっている[37]。エクソン1A、1B、および1Cは、クローン化されたヒトDNAに見られるものと61〜78%の相同性を示した[37、38]。代替プロモーターは発生および組織特異的な遺伝子調節に関連しており、α2タンパク質産物に検出された差異はないものの、6つのα2アイソフォームに関わっている可能性がある[37]。GABRA2のこれら50隣接領域のヌクレオチドシーケンス分析により、3つの代替プロモーターすべてがグアニン-リン酸-シトシンジヌクレオチド(GpC)に富む領域に配置され、3つのうち2つは典型的なTATAおよびCCAATシーケンスを欠いていることがわかった。ラットとヒトのGABRA2のシリコ比較(silico comparison)では、GABRA2に隣接する50領域に保存されたSp1サイトが確認された[29]。これは、ニューロン固有の転写因子Sp4 [31]により調節されることが実験で確認されている。
GABRA3遺伝子はX染色体に局在し、GABRB4遺伝子とクラスターを形成している[39]。マウスとヒトにおいて、cDNAはGABRA3と他のGABAARサブユニット遺伝子間に非常に似通ったイントロン-エクソン構造を示し、コアプロモーターにGAリピートを含む非常にユニークなプロモーター領域が存在する[39]。複数リピートは種によって異なりランダムであるように見え、一般に転写に与える影響は少ない[39,40]。GABRA3遺伝子にはいくつかのスタートサイトがあるように見えるが、プロモーターが複数あるというエビデンスはない[29]。GABRA4プロモーターの検査により示されたのは、Sp3およびSp4に結合する2つのSp結合部位が同定されているということであった。それは生体内でのプロモーター活性に重要である[41]。GABRA4プロモーターには、多くの種で共通して初期保存応答タンパク質(Egr)サイトが含まれており[42]、Egr3は発作後のα4サブユニット発現変化をレギュレーションすることが示されている[42]。Egrファミリーのメンバー、特にEgr1は、物質の使用後に線条体で誘発されることが示されている[43、44]。
他のGABAARサブユニットには、遺伝子発現の適切な時間的および空間的調節に役立つ代替プロモーターがある。 ヒトのGABRA5遺伝子には、ラットのホモログと類似する少なくとも3つの異なるエクソンが存在し、このプロモーターの進化的重要性が示唆されている[24]。α5の代替プロモーター配列は、他のGABAARサブユニットプロモーターの配列と似てはいないが、共通してTATAおよびCCAATボックスを欠いており、シトシン–リン酸–グアニンジヌクレオチド(CpG)に富む配列である[ 24]。GABRA5プロモーターには、Sp結合の推定部位、および活性化タンパク質2(AP2)モチーフが含まれる[29]。α6サブユニットを含むGABAARは、小脳に限定された高度に制御された発現を示し、50-UTRに関する研究がこのレギュレーションプロセスを明示している。とりわけ小脳顆粒細胞でのGABRA6遺伝子発現増強領域には、保存された核因子1(NF-1)結合部位が含まれていることがわかっている[45、46]。クロマチン免疫沈降は、NF-1がGABRA6プロモーターに結合し、NF1の削除が小脳顆粒細胞におけるα6の発現を劇的に減少させることを実証した[45]。
転写の50UTR調節に加えて、GABAARサブユニット遺伝子は、遺伝子本体への影響によっても調節される。たとえば、50UTRおよびGABRB3遺伝子には、メチルCpG結合タンパク質2(MeCP2)の結合部位が含まれている。MeCP2は5-メチルシトシンを認識し結合する、つまりメチル化の解読者である。MeCP2結合は、主にポリメラーゼの遅延による転写の抑制につながると考えられているが[47]、同時に活性化因子であることも示唆されている[48]。MECP2遺伝子の機能喪失変異は、神経発達障害のレット症候群を引き起こす[49、50]。レット症候群の患者の死後組織、および神経発達障害のエンジェルマン症候群の検査では、β3サブユニットの発現が低下していることが示されている[50、51]。この発見は、β3サブユニットがダウンレギュレートされたMeCP2ヌルマウスの小脳で確認されている。さらに、α1サブユニットが前頭皮質でダウンレギュレートされているのに対し、α2およびα4は
MeCP2ヌルモデルの腹外側髄質で減少した[51、52]。生体内実験では、MeCP2発現はGABAARα1サブユニット発現活性化が見られた[53]。レット症候群患者の組織およびモデルにおける複数のGABAARサブユニット発現パターンの調節は、MeCP2がGABAARサブユニット遺伝子のグローバルな転写レギュレーターである可能性があることを示唆している[53]。MeCP2のリン酸化を介して引き起こされるエピジェネティックなレギュレーションは、物質使用暴露後に遺伝子発現の変化に関与していることがわかっている[54]。
全体的に、複数のGABAARαサブユニット遺伝子は、GpCもしくはCpGリッチであり、標準的なTATAボックスを欠く50-UTRに隣接している(図2)。αサブユニット遺伝子は多くの場合、複数のプロモーター、開始部位を持ち、複数の転写因子のコンセンサス配列を含んでいるため、高度な制御が示唆される。βサブユニットとγサブユニットの遺伝子は同様の特徴を示しており、一部(β1、β3、γ1)には標準のTATAボックスがなく、種間で比較的よく保存されている。GABAARサブユニットの転写制御の解明は、GABAARサブユニット発現の空間的、時間的調節メカニズムの強力な仮説につながっている。しかしながら、いくつかの転写因子コンセンサスサイトの検証、および発現への機能的影響の実証には、さらに研究が必要である。それは成長期全体にわたる時間的調節および様々な刺激との関係にまで及ぶかもしれない。加えて、CREB、Egrファミリーメンバー、MeCP2などが物質使用に対する反応に関係していることは分かっているが、これらの要因が物質使用障害発症の原因となる不適応可塑性、そしてそれによるGABAAR発現調節がどのようなプロセスになるかまではわかっていない。
2.2. 翻訳
転写によるmRNAの産生に続いて、mRNAのタンパク質産物への翻訳が遺伝子発現のさらなるレギュレーションにつながることになる。mRNAの翻訳は、3'-UTRの長さや構造安定性、mRNA結合部位の量、3'-UTRにおけるアデニル酸-ウリジル酸(AU)要素などの機能だけでなく、さまざまなタンパク質の影響を受ける多段階プロセスである [55,56]。mRNAは、mRNAとさまざまなRNA結合タンパク質(RBP)から構成されるイボ核タンパク質粒子に組み込まれ、同族のRBPからのmRNAの放出により翻訳が開始される[57,58]。最近の研究では、非標準的なRNA結合ドメインを特定することにより、非常に多くのRBPが確認されてきている[59]。RBPのトランスクリプトーム全体の解明によって、ほとんどのRBP結合が転写物3’-UTRで発生することが明らかになった[60]。マウス3'-UTRomeと比較して、GABAARサブユニットはその3'-UTRが非常に長い[55]。そのことは、HEK細胞とヒトニューロンの翻訳活動の低下に関連し[55、61、62]、GABAARサブユニット発現に対する高度な制御を示唆している。3’-UTRは成長期にも長くなり、成熟したニューロンの翻訳調節が成長期にくらべてより増強されることを示している。 GABAARサブユニットタンパク質、scffoldタンパク質、および輸送タンパク質のmRNAは、RBPのターゲットとして識別されている(図2)[55]。
RBP Pumilio2(Pum2)は、標的mRNAの30-UTRの8ヌクレオチドコンセンサスシーケンスに結合する転写後レギュレーターである[63]。てんかんの特徴を持つPum2ノックダウンモデルでは、GABRA2 mRNAが2倍にアップレギュレートされた[63]。以前の研究では、Pum2がRNA崩壊を促進するデデニラーゼ複合体を動員することが示されている。これはPum2が存在しない場合、α2サブユニットのmRNAがより安定し、発現の増加につながる可能性があることを示唆している[63]。非POUドメインを含むオクタマー結合タンパク質(NONO)は、ショウジョウバエの行動/ヒトスプライシング(DBHS)ファミリーのタンパク質に由来するDNAおよびRNA結合タンパク質である。NONOをコードする遺伝子の変異は、人間の知的障害に関連している[64]。 NONOはニューロンのRNA輸送複合体のメンバーであり[57]、活動依存的な方法でシナプスに局在する[65]。NONOは多数の転写産物を調節することが示されているが、その3分の1がシナプスタンパク質である[64]。シナプスタンパク質発現に対するNONOの効果は、そのRNA結合ドメインによって調節されている[64]。 mRNAおよびα2サブユニットタンパク質の減少は、NONO遺伝子破壊を伴うマウスモデルの海馬で検出された[64]。これらの発見は、NONOがα2サブユニットのmRNAの安定性と翻訳の重要なプロモーターである可能性があることを示唆している。物質使用におけるPum2とNONOを調べた研究はないが、他のRBPが物質使用障害の発症に関係しているとされており[66]、翻訳調節に関する研究は非常に興味深いテーマとなっている。
フラジャイルX精神遅滞タンパク質(訳注:Fragile X mental retardation protein. FMRP)は、ユビキタスに発現するKH含有RBPであり、ポリリボソームと関連している[67]。FMRPは、リボソームに直接結合することによってmRNAの翻訳を阻害することが示され、リボソーム上のtRNAおよび翻訳伸長因子の結合を妨げる[68]。FMRPはまた、シナプス部位へのmRNAの輸送および局在化、ならびにシナプスタンパク質合成において一定の役割を果たすことが示唆されている[69]。一部推定ではあるが、FMRPが神経系遺伝子の約5%をターゲットとしていることが示唆されている。フラジャイルX精神遅滞1(FMR1)遺伝子の50-UTRのCCGトリヌクレオチドリピート拡張は、FRMPのサイレンシングを引き起こし、神経発達障害のフラジャイルX症候群を引き起こす[70]。FMRPノックアウトマウスでの複数の研究では、α1、β2、δサブユニットの発現のダウンレギュレーション[71]と、α1、3-5、β1-3、γ1、2、δサブユニットのmRNAの減少が領域的な特異性で示されている[72–74]。FMRPがサブユニット発現を調節するメカニズムは不明なままだが、これらの結果は、FMRPがGABAARの選択したサブユニットのトラフィッキングおよび/または翻訳の調節に関与していることを示唆している。最近のデータはまた、FMRPが物質への曝露によって誘発される可塑性の、負の調節因子として作用することが研究で示されている[54]。
マイクロRNA(miRNA)は、3’UTRのmRNAと結合し、遺伝子発現に影響を与える小さな非コードRNAである[75、76]。miRNAは、mRNA内の相補配列との塩基対合を介して翻訳を制御し、翻訳効率の低下、mRNAの不安定化、またはmRNA切断を引き起こすことによって遺伝子発現を抑制する。シリコ分析(silico analysis)では、複数GABAARサブユニットのmiRNA結合部位を予測できた[77]。これらの研究はまた、α1mRNAにおけるmiRNAの少なくとも6つの結合部位と、αサブユニットにおいてその結合部位数を予測できた[77]。これらの研究では、α2または3の結合部位は予測されず、α4および5の予測部位はmiRandaのみであった[77]。β1-3サブユニットのmRNAでは、複数のmiRNA結合部位が予測できた[77]。γ2サブユニットは2つのサイトを持つと予測できた[77]。最も一般的に発現されるサブユニットは、miRNA結合部位が最も多く予測され、発現が制限されたサブユニットはより少ないと考えられる。これはおそらく3'UTRが短いためであろう[77]。実験では、α1サブユニットをコードするmRNAは、miR-181aによって調節されることが示され、miR-181aのレベルが上昇すると、α1サブユニットの発現がダウンレギュレートされる[78]。miR-186、miR-24、またはmiR-375の発現は、α4発現をダウンレギュレートすることが示されている[79]。miR-203は、α5サブユニットをコードするmRNAのルシフェラーゼ活性報告30UTR活性を低下させることが示されている[80]。miRNAsがGABAARサブユニット発現のレギュレーターであるというエビデンスが積みあがっているが、詳細はまだ解明されていない。物質使用障害における遺伝子発現miRNA調節の役割を証拠づけるエビデンスも増えてきている[81–83]。これについては、以下で詳しく述べる。
3. 経験依存可塑性によるGABAA受容体サブユニット発現調節
外部および内部刺激に適応する能力は、神経系のユニークな特性である。脳は、シナプス接続とその応答メカニズムによりシグナル伝達を調節することで、生理学的および病理学的コンテキストに適応することが知られている。物質使用への応答および物質使用障害の発症に関与した、学習と記憶のダウンストリーム可塑性を制御するメカニズムがいくつかのエビデンスで証明されている。
シナプスが神経活動によって動的に調節されることは明らかになっている[84–87]。多くの研究が経験依存性興奮性シナプスの可塑性を調べてきたが、抑制性シナプスを調節する分子メカニズムについてはあまり知られていない[88,89]。感覚経験は哺乳類の脳の発達とそのリファインメントという、いくつかのプロセスをコントロールする。その多くは興奮性シナプスでのグルタミン酸放出に起因し、最終的にシナプスの強さと数の変化をもたらすことになる[84,86,88]。そしてそれは翻って、恒常性を維持するために、抑制性シナプスの数と強さが感覚入力と興奮性活動のレベルに影響される[88,90–92]。GABA作動性抑制の動的調節は、ネットワーク適応を可能にしながらホメオスタシスを維持するための不可欠な調節だというわけである[93]。
CREBはニューロンの可塑性のメカニズムの中心となることが明らかになっている。 CREBは、cAMP応答要素(CRE)として知られるDNA内の配列に結合する転写因子で、多くの遺伝子の50UTRに含まれている。cAMPの産生および細胞内Ca2 +の増加はキナーゼを活性化し、それは翻ってCREBをリン酸化して活性化させる。リン酸化CREBはCREに結合し、コアクチベーターCREB結合タンパク質(CBP)と相互作用して、ヒストンアセチルトランスフェラーゼをプロモーターに昇格させる。上記に述べたように、複数のGABAARサブユニット遺伝子には、プロモーター領域にCREB結合のためのコンセンサスサイトがある。CREB活性化の他のダウンストリームターゲットには、さらなる遺伝的調節へのゲートウェイとして機能する中間初期遺伝子(IEG)が含まれる。c-Fos、c-Myc、c-JunなどのIEG転写因子は、細胞の成長と分化のユビキタスレギュレーターとして広く知られている。神経系が刺激と経験によって絶えず調節されていることを考えると、多くのIEG転写因子がニューロンで識別され、特徴付けられていることは驚くことではない。活動調節細胞骨格関連タンパク質(Arc)とEgr-1は、他のIEG転写因子の中でも、感覚体験による神経活動の変化を反映することが示され、それらのアップレギュレーションが神経活動を決定するプロキシとして使用されている[94 、95]。
IEG脳由来神経栄養因子(BDNF)はCREBのダウンストリームターゲットであり、GABAARサブユニットの発現の重要な調節因子である。BDNFは、Janusキナーゼ(JAK)/シグナルトランスデューサーおよび転写活性化(STAT)経路を活性化することにより、α1サブユニット転写のCREB制御を駆動する。JAK / STAT経路はBDNFの発現を調節し、最終的に50UTRへのCREB / ICER結合を介してGABRA1転写を抑制する[32]。BDNFはまた、プロテインキナーゼc(PKC)/マイトジェン活性化プロテインキナーゼ(MAPK)経路を活性化し、Egr3のレベルを調節することにより、α4サブユニット発現を調節する。α1およびα4サブユニットに加えて、BDNFはα2、β2、β3、およびγ2サブユニット発現の調節にも関与している[96]。BDNFは、神経発達における形成要因であるだけでなく、学習と記憶[97]、そして物質使用[98,99]によって誘発される可塑性のファクターであることはよく知られている。
最近では、IEG転写因子ニューロンPASドメインタンパク質4(NPAS4)は、神経活動の抑制性シナプス接続ダウンストリームの重要なレギュレーターとして注目を集めている。 NPAS4は、膜の脱分極に応答してのみ発現し、経験依存である[88,89]。海馬錐体ニューロンでは、NPAS4は、頂端樹状突起から離れた細胞体への抑制性シナプス入力をバイアスすることで、抑制性シナプス数を調節する。細胞内ターゲティングに対するこの効果は、NPAS4標的遺伝子BDNFによって媒介されることが示されている[100]。感覚強化は、マウス海馬のCA1のNPAS4レベルを増加させ、抑制性コレシストキニン(CCK)バスケット細胞によって作られるシナプスを動員するが、パルブアルブミンバスケット細胞ではそうはならない[100]。CCKバスケット細胞からのコンタクト数を増やすと、GABAAR発現が増加し、錐体細胞の細胞体で豊富になるが、GABAARサブユニットのレギュレーションのメカニズムは解明されていない。
4. 物質使用によるGABAA受容体サブユニットの発現調節
物質使用を継続することで、脳の報酬システムに病的な可塑性を誘発し、正常な恒常性行動を強制的に薬物探索に変換する。広義には物質使用障害とは、有害であるにもかかわらず、違法であれ合法であれ、物質または薬物の使用を制御できないこととして定義されている。物質使用障害は、精神障害の診断および統計マニュアル(DSM-5)の第5版で、危険な使用、正しい使い方の遺棄、耐性、離脱症状、渇望など12の基準によって診断される[101]。3つ以上の基準を満たすことで薬物使用障害とみなされ、満たす基準が多ければ多いほど重症度が高い。耐性とは、物質使用が繰り返されることで連続した同用量に対する反応低下のことである。離脱症状とは、物質の突然の除去時に発生し、慢性的な過敏性、感情的な痛み、倦怠感、不快感、ストレス、自然の報酬に対する動機の喪失などがある[102]。物質使用の頻繁な使用は、主に腹側被蓋野(VTA)から側坐核(NAc)[103,104]の内側の殻までの中脳辺縁系経路のドーパミン(DA)シグナルを介してポジティブな強化を引き起こす。このプロセスは、乱用された物質の強化効果や、食品などの常態強化因に関係している[103,105]。物質使用を繰り返すことで、VTAのDAニューロンへの興奮性求心性神経を増強し、NAcにシナプス可塑性を引き起こす可能性がある[106]。回路の適応はまた、拡張された扁桃体のコルチコトロピン放出因子、ノルアドレナリンなどのストレスメディエーター、そしてNAcによる適応を通じて、長期薬物使用の突然の終了に対する嫌悪反応を引き起こす[103,105]。転写および転写後の遺伝子調節は、物質使用障害の発症に関連する脳機能の変化の大きな因子であると考えられている[55、107]。
DAergicシグナル伝達への直接作用がないにもかかわらず、GABAARを積極的に調節する薬物の多くは乱用されがちである。GABAARに対するGABAの作用は、ベンゾジアゼピン(BZ)、バルビツール酸塩、ステロイド、麻酔薬などの薬理学的および臨床によく使用されるさまざまな薬物によって調節されることがよく知られている[2]。一般にこれらの薬物は、GABAARの固有の部位でポジティブアロステリックモジュレーター(PAM)として機能し、GABA結合の効果に変化をもたらす[108]。BZは通常、αサブユニットとγサブユニット間の細胞外ドメインの特定のBZサイトに結合することにより、GABAARのPAMとして機能する[108]。GABAARは、プロポフォール、エトミデート、ハロタンなどの全身麻酔薬のターゲットでもあり、異なる麻酔薬は異なるサブユニット選択性と結合部位を持っている[108,109]。アルコールもまた、GABAARに作用すると考えられている。その作用は低用量(<30 mm)アルコール使用に対するGABA阻害Cl-電流の感受性に左右され、その変調メカニズムについてはいくつかの説がある[108,110–112]。多くの研究がGABAAR PAMについて調査してきたが、GABA作動性シグナルのポジティブモジュレーションがDA作動性報酬経路の活性化につながる機序、および物質使用障害の発症におけるGABA作動性シグナルの正確な役割については、いまだ多くの疑問が残っている。
4.1. ベンゾジアゼピン
GABAARはBZの主要な結合部位である。BZは不安、不眠症、てんかんなどの疾患の治療に広く使用される[113]。α(1、2、3、または5)とβおよびγサブユニットで構成されるGABAARは、BZ感受性受容体と見なされる[114]。 BZはイオンチャネルを開かせ流入を増加させることにより、GABAARのClコンダクタンスを変調する[115]。BZは非常に多く処方されていて、多くの副作用に関係しており耐性がつきやすく、依存症や離脱症状を引き起こし、乱用されることもある。耐性、依存、離脱の発生は、GABAARの発現、結合、機能の変化と関係している(表1に要約)。
表1.ベンゾジアゼピン(BZ)とアルコール:継続使用とその離脱によるGABAA受容体サブユニットレギュレーション(注釈:↓=ダウンレギュレーション、↑=アップレギュレーション)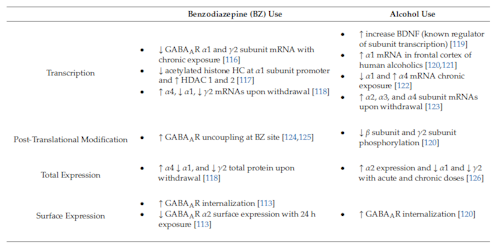
抗不安作用への耐性は、マウスにロラゼパムを1回投与した後48時間で示された[127]。耐性形成の根底にあるメカニズムはまだ完全には解明されていないが、BZ連用によって、ダウンストリームでのGABAARサブユニット遺伝子発現変化は実証されている。生体内でBZを慢性投与し、定量的リアルタイムPCRによってGABAARサブユニット発現を測定した結果、ロラゼパムを投与(14日間と28日間)した後に皮質α1サブユニットmRNAが50%の減少した[116]。対照的に、より少ない期間でのロラゼパム投与(1日、2日間、4日間、7日間、10日間)では、α1サブユニットmRNAを変更しなかった[116]。γ2サブユニットmRNAの減少も同様であった[116]。
エピジェネティックメカニズムもまた、耐性形成につながるGABAARサブタイプのダウンレギュレーションに関与している[ 117]。それはDNAメチル化、ヒストンのアセチル化とメチル化、クロマチン修飾およびその他のメカニズムを通じてよく理解できる。遺伝子発現のエピジェネティック調節は、物質使用障害を含む精神障害、ならびに耐性、依存、離脱症状など物質使用に対する神経系の適応に関与している[128]。部分的PAMイミダゼニルは、BZを慢性的に使用しても抗けいれん効果に対する耐性を形成せず、完全PAMで見られるようなα1サブユニットmRNAの発現低下にはつながらない[117]。部分的PAMがα1サブユニットの発現へ影響しないのは、GABRA1のアップストリームプロモーター領域でのヒストンのアセチル化の違いによるものであろう[117]。完全PAMであるジアゼパムは、プロモーターでアセチル化ヒストンH3の減少を示し、プロモーター領域でのMeCP2占有率を増加させた[117]。H3アセチル化の減少は、ヒストンデアセチラーゼ(HDAC)酵素HDAC1およびHDAC2の発現増加によるものであった[117]。クラスI HDACは、遺伝子のプロモーターで転写リプレッサーとして機能するMeCP2と複合体でしばしば見られる。これは、BZサイトで完全PAMの慢性投与によるα1サブユニットダウンレギュレーションのメカニズムを示唆している[117,129,130]。総アセチル化ヒストンH3タンパク質の評価により、ジアゼパムの長期使用は皮質のH3タンパク質レベルを低下させておらず、その低下はGABRA1のプロモーターに結合したものに特異的であった[117]。
さらに複数の研究により、GABAとBZの結合部位の脱共役のエビデンスが示されており、アロステリックな増強は結合親和性の変化なしに減少することがわかっている[113,131–133]。完全PAMであるジアゼパムまたは部分的PAM Ro 16 6028を投与すると、生体内ラットニューロンのGABAに対する受容体感受性が50%低下した[134]。初期の実験では、ニワトリ胚ニューロン培養を使用して、18時間で約34%の脱共役を引き起こし、それを介してBZフルラゼパムに対する耐性が急速に形成されることを示した[131]。さらに、BZサイトアンタゴニストのフルマゼニル、ピクロトキシン、GABAARチャネルブロッカー、またはL型電位依存性Ca2 +チャネルの阻害剤であるニフェジピンを使用することで、脱共役が防止された。このことは、特定の部位へのジアゼパムの結合と、それに続くGABAARの活性化のメカニズムを示している。
シナプス後GABAARの表面レベルはシナプス抑制の強さに関連しており、原形質膜へ/からの流入出トラフィックによって調節される。受容体の除去、分解、挿入、および分散はすべて動的に調節されることが示されている[136]。BZフルラゼパムによる治療後のGABAAR表面レベルを調べたところ、サブユニットの総量とともに、α2サブユニットを含むGABAAR表面発現の劇的な減少を示していた[113]。BZ治療は、α2を含むGABAARの挿入率とエンドサイトーシス率を変化させなかったが、分解を促した。これはリソソーム分解をブロックすることによって逆転した[113]。総GABAARレベルの欠損は、治療開始の24時間で受容体分解を引き起こし、耐性形成につながる一連の神経適応を作動させる。
4.1. アルコール
GABA作動性システムに作用する薬物と同様にアルコールもまた、抗不安、抗けいれん、鎮静催眠、認知機能障害、運動協調機能障害につながる特性をもっている。その背後にあるメカニズムには、GABAARへの直接的および間接的な影響、GABAの放出と合成の調節、内因性神経活性ステロイドの利用可能性[120]、および他の神経伝達物質系への作用である。アルコールへの暴露に関する研究が示しているのは、アルコール使用とアルコール使用障害の発症に関与するエピジェネティックメカニズムが存在するということである[137,138]。慢性的なアルコール使用の影響について、信頼できるバイオマーカーを用いてエピゲノム全体の関連研究を行われた[139]。それは13の異なるコホートにわたるアルコール消費レベルに関連したCpGサイトのメチル化の差異を調べたものである。そこでは144のCpGがアルコール消費に関連していること、およびGABAARおよびGABABRサブユニットのエピジェネティックな変化を発現した遺伝子は、免疫機能に関与する多くの遺伝子の発現レベルと有意に関連していた[139]。アルコール使用に最も関連するCpGのうち、cg04781796はGABAARδサブユニット(GABRD)に対してイントロニックなCpGアイランドにあり、cg09577455はGABABRサブユニット1(GABBR1)に対してイントロニックなCpGアイランドにある。どちらも免疫機能に関係している[139]。BZと同様に、HDAC活性の低下によって引き起こされるヒストンのアセチル化の変調はアルコールでも見られ、抗不安作用に関与していると考えられている[119]。CREBターゲット遺伝子BDNF、Arc、およびニューロペプチドYはすべて、急性アルコール曝露により扁桃体で増加し[36]、GABAARサブユニットのレギュレーションをもたらすカスケードを開始する可能性がある。
長期のアルコール連用は、さまざまな脳領域でGABAARサブユニットmRNAの発現とタンパク質レベルに異なる変化を引き起こすことが示されている(表1に要約)[120]。α2サブユニットのmRNAは、アルコール依存症患者の中枢扁桃体でダウンレギュレートされている[140]。ヒトのRT-PCR研究では、アルコール依存者の前頭皮質におけるα1発現の増加、および対照と比較した総αサブユニット濃度が示されている[120、121、141]。対照的に、げっ歯類では、慢性的なアルコール暴露でα1発現の減少とα4発現の増加が示されている[120、122、126]。慢性間欠エタノール(CIE)ラットモデルでは、α2を含むGABAARは、海馬、基底外側扁桃体、およびNAcでアップレギュレートされ、これらはすべて中毒調節に関与している[126]。カニクイザルをアルコールに慢性曝露させると、側底扁桃体のα2およびα3サブユニットのmRNAを減少させ、α1サブユニットmRNAを増加させ[142]、また別の皮質領域におけるGABAARサブユニットmRNAの発現に影響した[143]。α2、α4、β1、β3、γ1およびγ3サブユニットのmRNAは眼窩前頭皮質で大幅に減少し、β1、β2、γ1およびδサブユニットmRNAは背外側前頭前皮質で減少した[143]。
特定のGABAARサブユニットを欠くげっ歯類の変異体モデルは、アルコールに対する生理学的および行動的応答の変化を示す[144–149]。α1ヌル変異マウスはアルコールに対する強い拒否反応を示す[144]。α1およびβ2ヌル変異体は、アルコール鎮静効果が変化し、アルコール摂取後の生反射喪失期間が短くなる[145]。α1およびβ2ノックアウトマウスは、アルコール暴露による自発運動刺激効果の増加も示した[144、146、147]。α2のノックダウンは暴飲のような飲酒の減少も示した[149]。NAcシェルでのα4サブユニットの発現を低下させるために、ウイルス媒介mRNA干渉すると、低から中程度のアルコール消費量減少につながる。これは、α4サブユニットがアルコール嗜好に関与している可能性を示している[150]。δサブユニットを欠くマウスは、自発的なアルコール摂取を好まない[148]。δサブユニットノックアウトマウスは、慢性的なアルコール暴露後の離脱で起きる興奮性亢進も低下する[148]。動物モデルとヒトのアルコール使用障害集団における研究では、アルコール依存症家族の遺伝的原因と合わせて、GABAARαサブユニット発現調節がアルコール使用障害の原因であるとしている[151,152]。
miRNAおよびその他の非コードRNAは、経験依存のシナプス可塑性における物質使用障害の要因として俎上に上がっている[76,137]。アルコール使用障害のあるヒト被験者の前頭皮質組織で、miRNA分析により、約35のmiRNAのアップレギュレーションが明らかになった[153]。シナプス可塑性のメディエーターが、検出されたmiRNAの変化の主な要因であることが示唆された[153]。同様に、慢性的なアルコール暴露の動物モデルでは、100を超えるmRNAと30を超えるmiRNAの発現変化が、いくつかの領域特異性とともに検出された[154]。遺伝子オントロジーと経路分析は、他の細胞シグナル伝達経路の中でシナプス可塑性に関与していることを示唆した[154]。GABAARサブユニットは慢性アルコール曝露におけるmiRNA変化の主要なターゲットではないが、関連経路とBDNFなどの調節因子であると複数の研究で明らかになっている。
4.1. その他の依存物質
精神刺激薬はGABAARに直接バインドしないが、ニコチン[164,165]、コカイン[166–168]、およびアンフェタミン[169]などの使用は、GABAARサブユニット発現調節に関わっている。アルコール使用と同様に、GABRA4とGABRA2のいくつかの一塩基多型は、ニコチン依存症にリンクしている[164]。α2サブユニットは、VTAニューロンのToll様受容体4の活性化によりニコチン依存症を引き起こし、CREBの活性化および副腎皮質刺激ホルモン放出因子とチロシンヒドロキシラーゼのアップレギュレーションにつながると考えられている[165]。人間のコカイン中毒者の海馬からのサンプルでは、
GABRG2は、GABAAR関連タンパク質ゲフィリン(GPHN)をコードする遺伝子とともにダウンレギュレートされた[168]。GABRG2とGPHNは両方とも、アルコール嗜好ラットの海馬でアップレギュレーションされており、ニコチンの自己投与とコカイン探索行動の増加も示している[168]。他の研究では、GABRA2遺伝子の削除により、条件付き強化を促進するコカインの効果が無効になることが示されている[166]。動物モデルでは、コカインに対する感作によってα2サブユニットレベルは、NAcシェルでは減少するが、NAcコアでは減少しない[167]。メタンフェタミンへの感作だと、NAcコアとシェルでα2発現減少、尾状核で発現増加する[170]。メタンフェタミン感作ラットはまた、前頭前野のα3およびβ1サブユニットのmRNAのアップレギュレーションを示した[169]。精神刺激薬に加えアヘンへの曝露もまた、GABAARサブユニットの発現変化を引き起こす可能性があるという指摘もある[171]。遺伝子ファミリーのマイクロアレイ研究では、14日間のモルヒネ暴露パラダイムの間に、NAcの特定サブユニットの誘導と抑制と混合を示唆している[172]。モルヒネ暴露約8日間で、全体的に、αおよびβサブユニット発現はやや抑制されたが、γ、δ、およびεサブユニット発現は増強された[172]。
4.4. 離脱症状
上で説明したように、GABAAR発現と機能は物質の使用に応じて変化し、耐性と物質使用障害の発症の一因であることが示されている。依存物質からの離脱でもまた、GABAARサブユニットの発現と機能を変化させることも示されている[123]。離脱の影響を観察するために、培養中の海馬ニューロンをエタノールに5日間連続暴露し、その後非エタノール培地を3〜24時間与え、mRNAレベル、ニューロンの形態、GABAARの機能的および薬理学的応答を調べた。エタノール暴露のみでは、α1、α3、α4、α5、およびγ2サブユニットの2つのバリアントのmRNAが減少したが、α2mRNAレベルは変化しなかった[123]。しかしながら、離脱中、α2、α3、およびα4はすべて大幅に増加、アルコール除去後約3時間でピークに達し、一方α1およびγ2サブユニットは、エタノール除去後9〜12時間でベースラインに戻った[123]。離脱中の薬理反応を観察するために、アルコール依存症患者や実験動物で離脱症状軽減に効果のある培地で培養ニューロンを培養してみた。培地に含む化合物は、ジアゼパム、γ-ヒドロキシ酪酸、GABABRアゴニストのバクロフェンなどである。ジアゼパムとγヒドロキシ酪酸が、離脱によって誘発されるα2、α3、およびα4サブユニットの増加を軽減した[123]。完全PAMであるBZジアゼパムも、α1およびγ2サブユニットの減少とα4mRNAと総タンパク質の増加を示した[118]。興味深いことに、サブユニット発現変化をあまり引き起こさない部分的PAMイミダゼニルは、α4発現を大幅に増加させ、α1とγ2の発現を減少させた[118]。アルコールとBZからの離脱に加えて、アヘンの離脱でもまた、GABAARサブユニット発現変化が指摘されている。 モルヒネ耐性ラットでは、離脱により青斑核でのサブユニットmRNAのアップレギュレーションが誘発され[173]、マイクロアレイ研究ではNAcでのGABAARサブユニット遺伝子の広範なダウンレギュレーションが示唆された[172]。4〜18日の断酒で、α、β、γ、δ、およびεサブユニットの抑制が確認された[172]。以上の結果により、GABAARサブユニット発現変化が離脱可塑性の共通特徴であることを示唆している。
アルコールからの離脱は、CREBのリン酸化の低下、ヒストンH3およびH4アセチル化の低下につながることも示されている。また、扁桃体のBDNF、Arc、およびニューロペプチドYが減少する[36]。偏桃体は、アルコール離脱症状のひとつ不安症状に関わる脳の領域である。これらの変化は、アルコール急性暴露で起こるものとは逆であり、離脱時のHDAC活動増加によって引き起こされると考えられている[36]。ラットのHDAC活性を阻害すると、アルコール離脱によって誘発される痛覚過敏が減少しする。これは、離脱に起因するエピジェネティックな修飾が痛みの処理にも影響を与える可能性があることを示唆している[174]。ラットを慢性的にアルコール曝露させた後、長期にわたって断酒させると、前頭皮質のmiRNAレベル変化も見られた[175]。この研究では、約165個のmRNAの変化とともに、40を超えるラットmiRNAが前頭皮質で変化することがわかった[175]。miRNA–mRNA発現ペアリングを使用すると、88のmRNAを標的とする推定の33のmiRNAが明らかになり、それらの多くはBDNFを含むシナプスシグナル伝達の調節に関与していた[175]。
まとめると、これらのデータは、薬物使用によるダウンストリームへの影響によるGABAAR発現変化と、薬物からの離脱によるGABAAR発現変化を明確に裏付けている。さらに、GABAARサブユニットの発現変化は、GABA作動性システムに直接作用する物質への曝露だけでなく、精神刺激剤やアヘンなど他の高依存物質への曝露でも見られる。GABAARサブユニットの発現変化は、サブユニット、調査中の物質、実験的パラダイム、タイムポイント、および脳の領域によって異なり、複雑である。NAcやVTAなどの特定の脳領域の変化は薬物報酬と関連しているが、前頭前野などの皮質領域の変化は薬物探索と選択行動に関連している可能性がある。GABAAR発現変化に至るメカニズムは多様で、とはいってもいくつかの一般的なファクターが浮かび上がる。学習と記憶(CREBとBDNF)に反応する可塑性、神経発達の可塑性(MeCP2とFMRP)、に関わるメカニズムと要因である。複雑さはともあれ、GABAAR発現変化は、依存物質への曝露と離脱の繰り返しにより発生する不適応可塑性の一貫した特徴であると思われ、さらに詳細な研究が必要である。将来の研究は、曝露後の特定の時点における、複数の脳領域にわたるサブユニットの変化に注視した研究になるであろう。報酬と強化、動機付けと快楽の影響、ならびに物質使用の動機となったストレスと不安など、物質使用障害における環境的側面を調べることも重要である。
5. Conclusion
GABAARは位置と機能によりその発現が異なる、さまざまなサブユニットで構成された特殊な受容体である[2、3]。特定のサブユニット発現は、感覚経験、物質使用、そして神経変性に至るさまざまな刺激に応答して、柔軟に適応する形で発生する。 BZやアルコールなどの物質作用により、サブユニット選択を変化させ、GABAAR発現そのものを調節したり受容体表面を変性させたりする[113,116,117,120,122,126,155]。mRNAの転写調節は最も一般的な発現調節手段だが、個々のGABAARサブユニット遺伝子の転写を制御する因子は完全にはわかっていない。染色体上のクラスター化配置を考慮すれば、GABAARサブユニットは単一の遺伝子から進化したと考えられており、ほとんどのGABAARサブユニット遺伝子は、CpGが豊富な標準的なTATAボックスがない50-UTRに隣接している[1,23,24 ]。CREBとBDNFは、複数のシグナル伝達経路を介して、複数のGABAARサブユニット発現にとって重要であり、それらは相互作用するレギュレーターであると考えられている[96,176]。これらのタンパク質は、学習と記憶の主な調節因子として定着しており[34,97]、物質使用に伴う可塑性においても同様である[35,36,98,99]。
転写後調節もまた、GABAARサブユニット発現に関わる。αサブユニットをコードする遺伝子の3'UTRomeは、マウスの3'UTRomeよりも長いため、RBPおよびmiRNAの多くの結合部位を利用して、発現の翻訳制御を行うことができる[1]。GABAARサブユニットmRNAをタンパク質へ翻訳調整するRBPが少数確認されている。翻訳調整されるタンパク質には次のようなものがある。Pum2(訳注:Pumilioホモログ2) [63,177]、NONO(訳注:non-POU domain-containing octamer-binding protein。非POUドメインを含むオクタマー結合タンパク質) [64,65]およびFMRP(訳注:fragile X mental retardation protein。脆弱X精神遅滞タンパク質) [55,67–74]。miRNAの結合部位は、GABAARサブユニット遺伝子の3'UTRで複数確認されている。miRNAは3’UTRの経験依存な可塑性に関与しており、遺伝子発現変化によって発症した物質使用障害の原因候補として浮上している[75–77,137,138]。
BZとアルコールはGABAARに作用し、サブユニット発現変化を引き起こし、HDAC阻害によるヒストンのアセチル化の増加などエピジェネティックな変性を引き起こす[36、117、138、174]。アルコール使用障害および動物モデルで、複数のサブユニットの発現と調節の変化が確認されており、また、α2サブユニットも遺伝的にアルコール依存症と関係している[108,149,151,152]。α2サブユニットはアルコールの抗不安作用を媒介することが知られており、不安増加を特徴とする離脱時に、扁桃体でもアップレギュレーションされる[118,123,174]。精神刺激剤のようなその他一般的な依存物質もGABAARサブユニットの発現を変更し、死後人体および動物モデルを使った研究で確認されている[164–172]。α2サブユニットでRBP結合部位が確認されているが、発現に影響を及ぼす特定のmiRNAおよびRBPは、特に物質使用においてさらに調査する必要がある。GABAA受容体の発現調節の上流にある要因を特定することで、3’UTRをターゲットとする物質使用障害の具体的な治療法につながる可能性がある。さらに、薬物への急性および慢性曝露、そして離脱中に被った遺伝子発現レギュレーションの要因を解明できる可能性がある。
Author Contributions: J.S.B. およびR.M.H. による共同執筆。図:R.M.H. 表:J.S.B。
Funding: 掲載料は、UNLV大学図書館オープンアーティクルファンドによって支援されました。
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Steiger, J.L.; Russek, S.J. GABAA receptors: Building the bridge between subunit mRNAs, their promoters,
and cognate transcription factors. Pharmacol. Ther. 2004, 101, 259–281.
2. Sieghart, W.; Fuchs, K.; Tretter, V.; Ebert, V.; Jechlinger, M.; Höger, H.; Adamiker, D. Structure and subunit
composition of GABAA receptors. Neurochem. Int. 1999, 34, 379–385.
3. Backus, K.H.; Arigoni, M.; Drescher, U.; Scheurer, L.; Malherbe, P.; Möhler, H.; Benson, J.A. Stoichiometry of
a recombinant GABAA receptor deduced from mutation-induced rectification. Neuroreport 1993, 5, 285–288.
4. Laurie, D.J.; Wisden, W.; Seeburg, P.H. The distribution of thirteen GABAA receptor subunit mRNAs in the
rat brain. III. Embryonic and postnatal development. J. Neurosci. 1992, 12, 4151–4172.
5. Ganguly, K.; Schinder, A.F.; Wong, S.T.; Poo, M. GABA Itself Promotes the Developmental Switch of Neuronal
GABAergic Responses from Excitation to Inhibition. Cell 2001, 105, 521–532.
6. Wisden, W.; Laurie, D.J.; Monyer, H.; Seeburg, P.H. The Distribution of 13 GABA, Receptor Subunit mRNAs
in the Rat Brain. I. Telencephalon, Diencephalon, Mesencephalon. J. Neurosci. 1992, 12, 1040–1062.
7. Pirker, S.; Schwarzer, C.; Wieselthaler, A.; Sieghart, W.; Sperk, G. GABAA receptors: Immunocytochemical
distribution of 13 subunits in the adult rat brain. Neuroscience 2000, 101, 815–850.
8. Hörtnagl, H.; Tasan, R.O.; Wieselthaler, A.; Kirchmair, E.; Sieghart, W.; Sperk, G. Patterns of mRNA and
protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience 2013, 236, 345–372.
9. Wu, X.; Wu, Z.; Ning, G.; Guo, Y.; Ali, R.; Macdonald, R.L.; De Blas, A.L.; Luscher, B.; Chen, G.
GABAA receptor alpha subunits play a direct role in synaptic versus extrasynaptic targeting. J. Biol. Chem.
2012, 287, 27417–27430.
10. Körber, C.; Richter, A.; Kaiser, M.; Schlicksupp, A.; Mükusch, S.; Kuner, T.; Kirsch, J.; Kuhse, J. Effects of distinct
collybistin isoforms on the formation of GABAergic synapses in hippocampal neurons. Mol. Cell. Neurosci.
2012, 50, 250–259.
11. Panzanelli, P.; Gunn, B.G.; Schlatter, M.C.; Benke, D.; Tyagarajan, S.K.; Scheiffele, P.; Belelli, D.; Lambert, J.J.;
Rudolph, U.; Fritschy, J.-M. Distinct mechanisms regulate GABAA receptor and gephyrin clustering at
perisomatic and axo-axonic synapses on CA1 pyramidal cells. J. Physiol. 2011, 589, 4959–4980.
12. Kneussel, M.; Brandstätter, J.H.; Gasnier, B.; Feng, G.; Sanes, J.R.; Betz, H. Gephyrin-independent clustering
of postsynaptic GABA(A) receptor subtypes. Mol. Cell. Neurosci. 2001, 17, 973–982.
13. Eyre, M.D.; Renzi, M.; Farrant, M.; Nusser, Z. Setting the Time Course of Inhibitory Synaptic Currents by
Mixing Multiple GABAA Receptor Subunit Isoforms. J. Neurosci. 2012, 32, 5853–5867.
14. Bright, D.P.; Renzi, M.; Bartram, J.; McGee, T.P.; MacKenzie, G.; Hosie, A.M.; Farrant, M.; Brickley, S.G.
Profound desensitization by ambient GABA limits activation of δ-containing GABAA receptors during
spillover. Version 2. J. Neurosci. 2011, 31, 753–763.
15. Picton, A.J.; Fisher, J.L. Effect of the alpha subunit subtype on the macroscopic kinetic properties of
recombinant GABA(A) receptors. Brain Res. 2007, 1165, 40–49.
16. Bianchi, M.T.; Haas, K.F.; Macdonald, R.L. α1 and α6 subunits specify distinct desensitization, deactivation
and neurosteroid modulation of GABAA receptors containing the δ subunit. Neuropharmacology 2002,
43, 492–502.
17. Mortensen, M.; Patel, B.; Smart, T.G. GABA Potency at GABAA Receptors Found in Synaptic and Extrasynaptic
Zones. Front. Cell. Neurosci. 2012, 6, 1–10.
18. Schofield, C.M.; Huguenard, J.R. GABA Affinity Shapes IPSCs in Thalamic Nuclei. J. Neurosci. 2007,
27, 7954–7962.
19. Keramidas, A.; Harrison, N.L. The activation mechanism of α1β2γ2S and α3β3γ2S GABAA receptors.
J. Gen. Physiol. 2010, 135, 59–75.
20. Dixon, C.; Sah, P.; Lynch, J.W.; Keramidas, A. GABAA Receptor α and γ Subunits Shape Synaptic Currents
via Different Mechanisms. J. Biol. Chem. 2014, 289, 5399–5411.
21. Mizukami, K.; Grayson, D.R.; Ikonomovic, M.D.; Sheffield, R.; Armstrong, D.M. GABAA receptor β2 and β3
subunits mRNA in the hippocampal formation of aged human brain with Alzheimer-related neuropathology.
Mol. Brain Res. 1998, 56, 268–272.
22. Ali, N.J.; Olsen, R.W. Chronic benzodiazepine treatment of cells expressing recombinant GABAA receptors
uncouples allosteric binding: Studies on possible mechanisms. J. Neurochem. 2001, 79, 1100–1108.
23. Russek, S.J. Evolution of GABAA receptor diversity in the human genome. Gene 1999, 227, 213–222.
24. Brooks-Kayal, A.R.; Shumate, M.D.; Jin, H.; Lin, D.D.; Rikhter, T.Y.; Holloway, K.L.; Coulter, D.A. Human
Neuronal γ-Aminobutyric AcidA Receptors: Coordinated Subunit mRNA Expression and Functional
Correlates in Individual Dentate Granule Cells. J. Neurosci. 1999, 19, 8312–8318.
25. Buckle, V.J.; Fujita, N.; Ryder-Cook, A.S.; Derry, J.M.; Barnard, P.J.; Lebo, R.V.; Schofield, P.R.; Seeburg, P.H.;
Bateson, A.N.; Darlison, M.G.; et al. Chromosomal localization of GABAA receptor subunit genes:
Relationship to human genetic disease. Neuron 1989, 3, 647–654.
26. McLean, P.J.; Farb, D.H.; Russek, S.J. Mapping of the alpha 4 subunit gene (GABRA4) to human chromosome
4 defines an alpha 2-alpha 4-beta 1-gamma 1 gene cluster: Further evidence that modern GABAA receptor
gene clusters are derived from an ancestral cluster. Genomics 1995, 26, 580–586.
27. Wilcox, A.S.; Warrington, J.A.; Gardiner, K.; Berger, R.; Whiting, P.; Altherr, M.R.; Wasmuth, J.J.; Patterson, D.;
Sikela, J.M. Human chromosomal localization of genes encoding the gamma 1 and gamma 2 subunits of the
gamma-aminobutyric acid receptor indicates that members of this gene family are often clustered in the
genome. Proc. Natl. Acad. Sci. USA 1992, 89, 5857–5861.
28. Lemon, B.; Tjian, R. Orchestrated response: A symphony of transcription factors for gene control. Genes Dev.
2000, 14, 2551–2569.
29. Joyce, C.J. In silico comparative genomic analysis of GABAA receptor transcriptional regulation.
BMC Genomics 2007, 8, 203.
30. Bateson, A.N.; Ultsch, A.; Darlison, M.G. Isolation and sequence analysis of the chicken GABAA receptor
α1-subunit gene promoter. Gene 1995, 153, 243–247.
31. Nair, B.; Johar, K.; Priya, A.; Wong-Riley, M.T.T. Specificity protein 4 (Sp4) transcriptionally regulates
inhibitory GABAergic receptors in neurons. Biochim. Biophys. Acta 2016, 1863, 1–9.
32. Brooks-Kayal, A.R.; Russek, S.J. Regulation of GABAA Receptor Gene Expression and Epilepsy. In Jasper’s
Basic Mechanisms of the Epilepsies; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V.,
Eds.; National Center for Biotechnology Information: Bethesda, MD, USA, 2012.
33. Lonze, B.E.; Ginty, D.D. Function and regulation of CREB family transcription factors in the nervous system.
Neuron 2002, 35, 605–623.
34. Kandel, E.R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain
2012, 5, 14.
35. Carlezon, W.A., Jr.; Duman, R.S.; Nestler, E.J. The many faces of CREB. Trends Neurosci. 2005, 28, 436–445.
36. Pandey, S.C.; Kyzar, E.J.; Zhang, H. Epigenetic Basis of the Dark Side of Alcohol Addiction. Neuropharmacology
2017, 122, 74–84.
37. Fuchs, K.; Celepirovic, N. The 50
-flanking region of the rat GABAA receptor α2-subunit gene (Gabra2).
J. Neurochem. 2002, 82, 1512–1523.
38. Hadingham, K.L.; Wingrove, P.; Le Bourdelles, B.; Palmer, K.J.; I Ragan, C.; Whiting, P.J. Cloning of
cDNA sequences encoding human alpha 2 and alpha 3 gamma-aminobutyric acidA receptor subunits and
characterization of the benzodiazepine pharmacology of recombinant alpha 1-, alpha 2-, alpha 3-, and alpha
5-containing human gamma-aminobutyric acidA receptors. Mol. Pharmacol. 1993, 43, 970–975.
39. Mu, W.; Burt, D.R. The mouse GABAA receptor α3 subunit gene and promoter. Mol. Brain Res. 1999,
73, 172–180.
40. De Groen, P.C.; Eggen, B.J.; Gispen, W.H.; Schotman, P.; Schrama, L.H. Cloning and promoter analysis of the
human B-50/GAP-43 gene. J. Mol. Neurosci. 1995, 6, 109–119.
41. Ma, L.; Song, L.; Radoi, G.E.; Harrison, N.L. Transcriptional regulation of the mouse gene encoding the
alpha-4 subunit of the GABAA receptor. J. Biol. Chem. 2004, 279, 40451–40461.
42. Roberts, D.S.; Raol, Y.H.; Bandyopadhyay, S.; Lund, I.V.; Budreck, E.C.; Passini, M.J.; Wolfe, J.H.;
Brooks-Kayal, A.R.; Russek, S.J. Egr3 stimulation of GABRA4 promoter activity as a mechanism for
seizure-induced up-regulation of GABA(A) receptor alpha4 subunit expression. Proc. Natl. Acad. Sci. USA
2005, 102, 11894–11899.
43. Bhat, R.V.; Cole, A.J.; Baraban, J.M. Role of monoamine systems in activation of zif268 by cocaine.
J. Psychiatry Neurosci. 1992, 17, 94–102.
44. Blackwood, C.A.; McCoy, M.T.; Ladenheim, B.; Cadet, J.L. Escalated Oxycodone Self-Administration and
Punishment: Differential Expression of Opioid Receptors and Immediate Early Genes in the Rat Dorsal
Striatum and Prefrontal Cortex. Front. Neurosci. 2019, 13, 1392.
45. Wang, W.; Stock, R.E.; Gronostajski, R.M.; Wong, Y.W.; Schachner, M.; Kilpatrick, D.L. A role for nuclear factor
I in the intrinsic control of cerebellar granule neuron gene expression. J. Biol. Chem. 2004, 279, 53491–53497.
46. McLean, P.J.; Shpektor, D.; Bandyopadhyay, S.; Russek, S.J.; Farb, D.H. A minimal promoter for the GABA(A)
receptor alpha6-subunit gene controls tissue specificity. J. Neurochem. 2000, 74, 1858–1869.
47. Cholewa-Waclaw, J.; Shah, R.; Webb, S.; Chhatbar, K.; Ramsahoye, B.; Pusch, O.; Yu, M.; Greulich, P.;
Waclaw, B.; Bird, A. Quantitative modelling predicts the impact of DNA methylation on RNA polymerase II
traffic. Proc. Natl. Acad. Sci. USA 2019, 116, 14995–15000.
48. Chahrour, M.; Jung, S.Y.; Shaw, C.A.; Zhou, X.; Wong, S.T.C.; Qin, J.; Zoghbi, H.Y. MeCP2, a Key Contributor
to Neurological Disease, Activates and Represses Transcription. Science 2008, 320, 1224–1229.
49. Kozinetz, C.A.; Skender, M.L.; Macnaughton, N.; Almes, M.J.; Schultz, R.J.; Percy, A.K.; Glaze, D.G.
Epidemiology of Rett syndrome: A population-based registry. Pediatrics 1993, 91, 445–450.
50. Zhang, Z.-W.; Zak, J.D.; Liu, H. MeCP2 Is Required for Normal Development of GABAergic Circuits in the
Thalamus. J. Neurophysiol. 2010, 103, 2470–2481.
51. Samaco, R.C.; Hogart, A.; LaSalle, J.M. Epigenetic overlap in autism-spectrum neurodevelopmental disorders:
MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 2005, 14, 483–492.
52. Medrihan, L.; Tantalaki, E.; Aramuni, G.; Sargsyan, V.; Dudanova, I.; Missler, M.; Zhang, W. Early defects of
GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol. 2008,
99, 112–121.
53. Oyarzabal, A.; Xiol, C.; Castells, A.A.; Grau, C.; O’Callaghan, M.; Fernández, G.; Alcántara, S.; Pineda, M.;
Armstrong, J.; Altafaj, X.; et al. Comprehensive Analysis of GABAA-A1R Developmental Alterations in Rett
Syndrome: Setting the Focus for Therapeutic Targets in the Time Frame of the Disease. Int. J. Mol. Sci. 2020,
21, 518.
54. Rothwell, P.E. Autism Spectrum Disorders and Drug Addiction: Common Pathways, Common Molecules,
Distinct Disorders? Front Neurosci. 2016, 10, 20.
55. Schieweck, R.; Kiebler, M.A. Posttranscriptional Gene Regulation of the GABA Receptor to Control Neuronal
Inhibition. Front. Mol. Neurosci. 2019, 12, 152.
56. Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The Mechanism of Eukaryotic Translation Initiation and Principles
of its Regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127.
57. Kanai, Y.; Dohmae, N.; Hirokawa, N. Kinesin Transports RNA: Isolation and Characterization of an
RNA-Transporting Granule. Neuron 2004, 43, 513–525.
58. Fritzsche, R.; Karra, D.; Bennett, K.L.; Ang, F.Y.; Heraud-Farlow, J.E.; Tolino, M.; Doyle, M.; Bauer, K.E.;
Thomas, S.; Planyavsky, M.; et al. Interactome of Two Diverse RNA Granules Links mRNA Localization to
Translational Repression in Neurons. Cell Rep. 2013, 5, 1749–1762.
59. Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev.
Mol. Cell Biol. 2018, 19, 327–341.
60. Andreassi, C.; Riccio, A. To localize or not to localize: mRNA fate is in 30UTR ends. Trends Cell Biol. 2009,
19, 465–474.
61. Floor, S.N.; Doudna, J.A. Tunable protein synthesis by transcript isoforms in human cells. Elife 2016, 5, e10921.
62. Blair, J.D.; Hockemeyer, D.; Doudna, J.A.; Bateup, H.S.; Floor, S.N. Widespread translational remodeling
during human neuronal differentiation. Cell Rep. 2017, 21, 2005–2016.
63. Follwaczny, P.; Schieweck, R.; Riedemann, T.; Demleitner, A.; Straub, T.; Klemm, A.H.; Bilban, M.; Sutor, B.;
Popper, B.; Kiebler, M. Pumilio2-deficient mice show a predisposition for epilepsy. Dis. Model. Mech. 2017,
10, 1333–1342.
64. Mircsof, D.; The DDD Study; Langouet, M.; Rio, M.; Moutton, S.; Siquier-Pernet, K.; Bole-Feysot, C.;
Cagnard, N.; Nitschké, P.; Gaspar, L.; et al. Mutations in NONO lead to syndromic intellectual disability and
inhibitory synaptic defects. Nat. Neurosci. 2015, 18, 1731–1736.
65. Zhang, G.; Neubert, T.A.; Jordan, B.A. RNA Binding Proteins Accumulate at the Postsynaptic Density with
Synaptic Activity. J. Neurosci. 2012, 32, 599–609.
66. Bryant, C.D.; Yazdani, N. RNA-binding proteins, neural development and the addictions. Genes Brain Behav.
2016, 15, 169–186.
67. Gantois, I.; Vandesompele, J.; Speleman, F.; Reyniers, E.; D’Hooge, R.; Severijnen, L.-A.; Willemsen, R.;
Tassone, F.; Kooy, R.F. Expression profiling suggests underexpression of the GABAA receptor subunit δ in
the fragile X knockout mouse model. Neurobiol. Dis. 2006, 21, 346–357.
68. Chen, E.; Sharma, M.R.; Shi, X.; Agrawal, R.K.; Joseph, S. Fragile X Mental Retardation Protein Regulates
Translation by Binding Directly to the Ribosome. Mol. Cell 2014, 54, 407–417.
69. Antar, L.N.; Dictenberg, J.B.; Plociniak, M.; Afroz, R.; Bassell, G.J. Localization of FMRP-associated mRNA
granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons.
Genes Brain Behav. 2005, 4, 350–359.
70. Liu, B.; Li, Y.; Stackpole, E.E.; Novak, A.; Gao, Y.; Zhao, Y.; Zhao, X.; Richter, J.D. Regulatory discrimination
of mRNAs by FMRP controls mouse adult neural stem cell differentiation. Proc. Natl. Acad. Sci. USA 2018,
115, E11397–E11405.
71. Adusei, D.C.; Pacey, L.K.K.; Chen, D.; Hampson, D.R. Early developmental alterations in GABAergic protein
expression in fragile X knockout mice. Neuropharmacology 2010, 59, 167–171.
72. D’Hulst, C.; De Geest, N.; Reeve, S.P.; Van Dam, D.; De Deyn, P.P.; Hassan, B.A.; Kooy, R.F. Decreased
expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006, 1121, 238–245.
73. Hong, A.; Zhang, A.; Ke, Y.; El Idrissi, A.; Shen, C.-H. Downregulation of GABA(A) β subunits is
transcriptionally controlled by Fmr1p. J. Mol. Neurosci. 2012, 46, 272–275.
74. Curia, G.; Papouin, T.; Séguéla, P.; Avoli, M. Downregulation of tonic GABAergic inhibition in a mouse
model of fragile X syndrome. Cereb. Cortex 2009, 19, 1515–1520.
75. Bushati, N.; Cohen, S.M. microRNA Functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205.
76. Sartor, G.C.; St. Laurent, G.; Wahlestedt, C. The Emerging Role of Non-Coding RNAs in Drug Addiction.
Front. Genet. 2012, 3, 106.
77. Zhao, C.; Huang, C.; Weng, T.; Xiao, X.; Ma, H.; Liu, L. Computational prediction of MicroRNAs targeting
GABA receptors and experimental verification of miR-181, miR-216 and miR-203 targets in GABA-A receptor.
BMC Res. Notes 2012, 5, 91.
78. Sengupta, J.; Pochiraju, S.; Pochiraju, S.; Kannampalli, P.; Bruckert, M.; Addya, S.; Yadav, P.; Miranda, A.;
Shaker, R.; Banerjee, B. MicroRNA-mediated GABAAα-1 receptor subunit downregulation in adult spinal
cord following neonatal cystitis-induced chronic visceral pain in rats. Pain 2013, 154, 59–70.
79. Bekdash, R.A.; Harrison, N.L. Downregulation of Gabra4 expression during alcohol withdrawal is mediated
by specific microRNAs in cultured mouse cortical neurons. Brain Behav. 2015, 5, e00355.
80. Janeczek, P.; Colson, N.; Dodd, P.R.; Lewohl, J.M. Sex Differences in the Expression of the α5 Subunit of
the GABAA Receptor in Alcoholics with and without Cirrhosis of the Liver. Alcohol. Clin. Exp. Res. 2020,
44, 423–434.
81. Bali, P.; Kenny, P.J. MicroRNAs and Drug Addiction. Front. Genet. 2013, 4, 335–344.
82. Li, M.D.; van der Vaart, A.D. MicroRNAs in addiction: Adaptation’s middlemen? Mol. Psychiatry 2011, 16,
1159–1168.
83. Smith, A.C.W.; Kenny, P.J. MicroRNAs regulate synaptic plasticity underlying drug addiction.
Genes Brain Behav. 2018, 17, e12424.
84. Zito, K.; Svoboda, K. Activity-Dependent Synaptogenesis in the Adult Mammalian Cortex. Neuron 2002,
35, 1015–1017.
85. Wong, R.O.L.; Ghosh, A. Activity-dependent regulation of dendritic growth and patterning.
Nat. Rev. Neurosci. 2002, 3, 803–812.
86. Spitzer, N.C. Electrical activity in early neuronal development. Nature 2006, 444, 707–712.
87. Katz, L.C.; Shatz, C.J. Synaptic Activity and the Construction of Cortical Circuits. Science 1996, 274, 1133–1138.
88. Lin, Y.; Bloodgood, B.L.; Hauser, J.L.; Lapan, A.D.; Koon, A.C.; Kim, T.-K.; Hu, L.S.; Malik, A.N.;
Greenberg, M.E. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature
2008, 455, 1198–1204.
89. Hartzell, A.L.; Martyniuk, K.M.; Brigidi, G.S.; Heinz, D.A.; Djaja, N.A.; Payne, A.; Bloodgood, B.L.
NPAS4 recruits CCK basket cell synapses and enhances cannabinoid-sensitive inhibition in the mouse
hippocampus. eLife 2018, 7, e35927.
90. Benevento, L.A.; Bakkum, B.W.; Cohen, R.S. gamma-Aminobutyric acid and somatostatin immunoreactivity
in the visual cortex of normal and dark-reared rats. Brain Res. 1995, 689, 172–182.
91. Hensch, T.K. Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 2005, 6, 877–888.
92. Marty, S.; Wehrlé, R.; Sotelo, C. Neuronal Activity and Brain-Derived Neurotrophic Factor Regulate the
Density of Inhibitory Synapses in Organotypic Slice Cultures of Postnatal Hippocampus. J. Neurosci. 2000,
20, 8087–8095.
93. Turrigiano, G.G.; Nelson, S.B. Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci.
2004, 5, 97–107.
94. Renier, N.; Adams, E.L.; Kirst, C.; Wu, Z.; Azevedo, R.; Kohl, J.; Autry, A.E.; Kadiri, L.; Venkataraju, K.U.;
Zhou, Y.; et al. Mapping of Brain Activity by Automated Volume Analysis of Immediate Early Genes. Cell
2016, 165, 1789–1802.
95. Bullitt, E. Expression of c-fos-like protein as a marker for neuronal activity following noxious stimulation in
the rat. J. Comp. Neurol. 1990, 296, 517–530.
96. Roberts, D.S.; Hu, Y.; Lund, I.V.; Brooks-Kayal, A.R.; Russek, S.J. Brain-derived Neurotrophic Factor
(BDNF)-induced Synthesis of Early Growth Response Factor 3 (Egr3) Controls the Levels of Type A GABA
Receptorα4 Subunits in Hippocampal Neurons. J. Biol. Chem. 2006, 281, 29431–29435.
97. Cunha, C.; Brambilla, R.; Thomas, K.L. A Simple Role for BDNF in Learning and Memory?
Front. Mol. Neurosci. 2010, 3, 1.
98. Logrip, M.L.; Barak, S.; Warnault, V.; Ron, D. Corticostriatal BDNF and alcohol addiction. Brain Res. 2015,
1628, 60–67.
99. Koskela, M.; Bäck, S.; Voikar, V.; Richie, C.T.; Domanskyi, A.; Harvey, B.K.; Airavaara, M. Update of
neurotrophic factors in neurobiology of addiction and future directions. Neurobiol. Dis. 2017, 97, 189–200.
100. Bloodgood, B.L.; Sharma, N.; Browne, H.A.; Trepman, A.Z.; Greenberg, M.E. The activity-dependent
transcription factor NPAS4 regulates domain-specific inhibition. Nature 2013, 503, 121–125.
101. Hasin, D.S.; O’Brien, C.P.; Auriacombe, M.; Borges, G.; Bucholz, K.; Budney, A.; Compton, W.M.; Crowley, T.;
Ling, W.; Petry, N.M.; et al. DSM-5 Criteria for Substance Use Disorders: Recommendations and Rationale.
Am. J. Psychiatry 2013, 170, 834–851.
102. Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016,
3, 760–773.
103. Lüscher, C.; Robbins, T.W.; Everitt, B.J. The transition to compulsion in addiction. Nat. Rev. Neurosci. 2020,
21, 247–263.
104. Sanchis-Segura, C.; Spanagel, R. Behavioural assessment of drug reinforcement and addictive features in
rodents: An overview. Addict Biol. 2006, 11, 2–38.
105. Koob, G.F.; Volkow, N.D. Neurocircuitry of Addiction. Neuropsychopharmacol 2010, 35, 217–238.
106. Mameli, M.; Halbout, B.; Creton, C.; Engblom, D.; Parkitna, J.R.; Spanagel, R.; Lüscher, C. Cocaine-evoked
synaptic plasticity: Persistence in the VTA triggers adaptations in the NAc. Nat. Neurosci. 2009, 12, 1036–1041.
107. Bali, P.; Kenny, P.J. Transcriptional mechanisms of drug addiction. Dialogues Clin. Neurosci. 2019, 21, 379–387.
108. Olsen, R.W. GABAA Receptor: Positive and Negative Allosteric Modulators. Neuropharmacology 2018,
136, 10–22.
109. Forman, S.A.; Miller, K.W. Mapping General Anesthetic Sites in Heteromeric Gamma-Aminobutyric Acid
Type A Receptors Reveals a Potential For Targeting Receptor Subtypes. Anesth. Analg. 2016, 123, 1263–1273.
110. Centanni, S.W.; Burnett, E.J.; Trantham-Davidson, H.; Chandler, L.J. Loss of δ-GABAA receptor-mediated
tonic currents in the adult prelimbic cortex following adolescent alcohol exposure. Addict Biol. 2017,
22, 616–628.
111. Wei, W.; Faria, L.C.; Mody, I. Low ethanol concentrations selectively augment the tonic inhibition mediated
by delta subunit-containing GABAA receptors in hippocampal neurons. J. Neurosci. 2004, 24, 8379–8382.
112. Herman, M.; Roberto, M. Cell type-specific tonic GABA signaling in the rat central amygdala is selectively
altered by acute and chronic ethanol. Addict. Biol. 2016, 21, 72–86.
113. Jacob, T.C.; Michels, G.; Silayeva, L.; Haydon, J.; Succol, F.; Moss, S.J. Benzodiazepine treatment induces
subtype-specific changes in GABAA receptor trafficking and decreases synaptic inhibition. Proc. Natl. Acad.
Sci. USA 2012, 109, 18595–18600.
114. Rudolph, U.; Möhler, H. Analysis of GABAA receptor function and dissection of the pharmacology of
benzodiazepines and general anesthetics through mouse genetics. Annu. Rev. Pharmacol. Toxicol. 2004,
44, 475–498.
115. Poncer, J.-C.; Dürr, R.; Gähwiler, B.H.; Thompson, S.M. Modulation of Synaptic GABAA Receptor Function
by Benzodiazepines in Area CA3 of Rat Hippocampal Slice Cultures. Neuropharmacology 1996, 35, 1169–1179.
116. Kang, I.; Miller, L.G. Decreased GABAA receptor subunit mRNA concentrations following chronic lorazepam
administration. Br. J. Pharmacol. 1991, 103, 1285–1287.
117. Auta, J.; Gatta, E.; Davis, J.M.; Pandey, S.C.; Guidotti, A. Potential role for histone deacetylation in chronic
diazepam-induced downregulation of α1-GABAA receptor subunit expression. Pharmacol. Res. Perspect.
2018, 6, e00416.
118. Follesa, P.; Cagetti, E.; Mancuso, L.; Biggio, F.; Manca, A.; Maciocco, E.; Massa, F.; Desole, M.S.; Carta, M.;
Busonero, F.; et al. Increase in expression of the GABA(A) receptor alpha(4) subunit gene induced by
withdrawal of, but not by long-term treatment with, benzodiazepine full or partial agonists. Brain Res. Mol.
Brain Res. 2001, 92, 138–148.
119. Pandey, S.C.; Ugale, R.; Zhang, H.; Tang, L.; Prakash, A. Brain Chromatin Remodeling: A Novel Mechanism
of Alcoholism. J. Neurosci. 2008, 28, 3729–3737.
120. Kumar, S.; Porcu, P.; Werner, D.F.; Matthews, U.B.; Diaz-Granados, J.L.; Helfand, R.S.; Morrow, A.L. The role
of GABAA receptors in the acute and chronic effects of ethanol: A decade of progress. Psychopharmacology
2009, 205, 529.
121. Lewohl, J.M.; Crane, D.I.; Dodd, P.R. Expression of the alpha 1, alpha 2 and alpha 3 isoforms of the GABAA
receptor in human alcoholic brain. Brain Res. 1997, 751, 102–112.
122. Matthews, D.B.; Devaud, L.L.; Fritschy, J.M.; Sieghart, W.; Morrow, A.L. Differential regulation of GABA(A)
receptor gene expression by ethanol in the rat hippocampus versus cerebral cortex. J. Neurochem. 1998,
70, 1160–1166.
123. Sanna, E.; Mosallino, M.C.; Busonero, F.; Talani, G.; Tranquilli, S.; Mameli, M.; Spiga, S.; Follesa, P.; Biggio, G.
Changes in GABAA Receptor Gene Expression Associated with Selective Alterations in Receptor Function
and Pharmacology after Ethanol Withdrawal. J. Neurosci. 2003, 23, 11711–11724.
124. Gutiérrez, M.L.; Ferreri, M.C.; Gravielle, M.C. GABA-induced uncoupling of GABA/benzodiazepine site
interactions is mediated by increased GABAA receptor internalization and associated with a change in
subunit composition. Neuroscience 2014, 257, 119–129.
125. Prasad, A.; Reynolds, J.N. Uncoupling of GABA-benzodiazepine receptors in chick cerebral cortical neurons
requires co-activation of both receptor sites. Brain Res. 1992, 591, 327–331.
126. Lindemeyer, A.K.; Shen, Y.; Yazdani, F.; Shao, X.M.; Spigelman, I.; Davies, D.L.; Olsen, R.W.; Liang, J.
α2 Subunit—Containing GABAA Receptor Subtypes Are Upregulated and Contribute to Alcohol-Induced
Functional Plasticity in the Rat Hippocampus. Mol. Pharmacol. 2017, 92, 101–112.
127. File, S.E.; Wilks, L.J.; Mabbutt, P.S. Withdrawal, tolerance and sensitization after a single dose of lorazepam.
Pharmacol. Biochem. Behav. 1988, 31, 937–940.
128. Berkel, T.D.M.; Pandey, S.C. Emerging Role of Epigenetic Mechanisms in Alcohol Addiction. Alcohol. Clin.
Exp. Res. 2017, 41, 666–680.
129. Nott, A.; Cheng, J.; Gao, F.; Lin, Y.-T.; Gjoneska, E.; Ko, T.; Minhas, P.; Zamudio, A.V.; Meng, J.; Zhang, F.; et al.
Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat. Neurosci. 2016,
19, 1497–1505.
130. Tesone-Coelho, C.; Varela, P.; Escosteguy-Neto, J.C.; Cavarsan, C.; Mello, L.E.; Santos-Junior, J.G. Effects of
ethanol on hippocampal neurogenesis depend on the conditioned appetitive response. Addict. Biol. 2013,
18, 774–785.
131. Roca, D.J.; Rozenberg, I.; Farrant, M.; Farb, D.H. Chronic agonist exposure induces down-regulation and
allosteric uncoupling of the gamma-aminobutyric acid/benzodiazepine receptor complex. Mol. Pharmacol.
1990, 37, 37–43.
132. Holt, R.A.; Bateson, A.N.; Martin, I.L. Decreased GABA Enhancement of Benzodiazepine Binding after a
Single Dose of Diazepam. J. Neurochem. 1999, 72, 2219–2222.
133. Wong, G.; Lyon, T.; Skolnick, P. Chronic exposure to benzodiazepine receptor ligands uncouples the
gamma-aminobutyric acid type A receptor in WSS-1 cells. Mol. Pharmacol. 1994, 46, 1056–1062.
134. Hernandez, T.D.; Heninger, C.; Wilson, M.A.; Gallager, D.W. Relationship of agonist efficacy to changes
in GABA sensitivity and anticonvulsant tolerance following chronic benzodiazepine ligand exposure.
Eur. J. Pharmacol. 1989, 170, 145–155.
135. Foitzick, M.F.; Medina, N.B.; Iglesias García, L.C.; Gravielle, M.C. Benzodiazepine exposure induces
transcriptional down-regulation of GABAA receptor α1 subunit gene via L-type voltage-gated calcium
channel activation in rat cerebrocortical neurons. Neurosci. Lett. 2020, 721, 134801.
136. Jacob, T.C.; Moss, S.J.; Jurd, R. GABAA receptor trafficking and its role in the dynamic modulation of
neuronal inhibition. Nat. Rev. Neurosci. 2008, 9, 331–343.
137. Feng, J.; Nestler, E.J. Epigenetic Mechanisms of Drug Addiction. Curr. Opin. Neurobiol. 2013, 23, 521–528.
138. Starkman, B.G.; Sakharkar, A.J.; Pandey, S.C. Epigenetics—Beyond the Genome in Alcoholism. Alcohol. Res.
2012, 34, 293–305.
139. Liu, C.; Marioni, R.E.; Hedman, Å.K.; Pfeiffer, L.; Tsai, P.-C.; Reynolds, L.M.; Just, A.C.; Duan, Q.; Boer, C.G.;
Tanaka, T.; et al. A DNA methylation biomarker of alcohol consumption. Mol. Psychiatry 2018, 23, 422–433.
140. Jin, Z.; Bhandage, A.K.; Bazov, I.; Kononenko, O.; Bakalkin, G.; Korpi, E.R.; Birnir, B. Expression of
specific ionotropic glutamate and GABA-A receptor subunits is decreased in central amygdala of alcoholics.
Front. Cell. Neurosci. 2014, 8, 288.
141. Mitsuyama, H.; Little, K.Y.; Sieghart, W.; Devaud, L.L.; Morrow, A.L. GABA(A) receptor alpha1, alpha4,
and beta3 subunit mRNA and protein expression in the frontal cortex of human alcoholics. Alcohol. Clin.
Exp. Res. 1998, 22, 815–822.
142. Floyd, D.W.; Friedman, D.P.; Daunais, J.B.; Pierre, P.J.; Grant, K.A.; McCool, B.A. Long-Term Ethanol
Self-Administration by Cynomolgus Macaques Alters the Pharmacology and Expression of GABAA
Receptors in Basolateral Amygdala. J. Pharmacol. Exp. Ther. 2004, 311, 1071–1079.
143. Hemby, S.E.; O’Connor, J.A.; Acosta, G.; Floyd, D.; Anderson, N.; McCool, B.A.; Friedman, D.; Grant, K.A.
Ethanol-Induced Regulation of GABAA Subunit mRNAs in Prefrontal Fields of Cynomolgus Monkeys.
Alcohol. Clin. Exp. Res. 2006, 30, 1978–1985.
144. Blednov, Y.A.; Walker, D.; Alva, H.; Creech, K.; Findlay, G.; Harris, R.A. GABAA Receptor α1 and β2 Subunit
Null Mutant Mice: Behavioral Responses to Ethanol. J. Pharmacol. Exp. Ther. 2003, 305, 854–863.
145. Blednov, Y.A.; Jung, S.; Alva, H.; Wallace, D.; Rosahl, T.; Whiting, P.-J.; Harris, R.A. Deletion of the α1 or β2
Subunit of GABAAReceptors Reduces Actions of Alcohol and Other Drugs. J. Pharmacol. Exp. Ther. 2003,
304, 30–36.
146. Boehm, S.L.; Ponomarev, I.; Jennings, A.W.; Whiting, P.J.; Rosahl, T.W.; Garrett, E.M.; Blednov, Y.A.;
Harris, R.A. γ-Aminobutyric acid A receptor subunit mutant mice: New perspectives on alcohol actions.
Biochem. Pharmacol. 2004, 68, 1581–1602.
147. June, H.L.; Foster, K.L.; Eiler, W.J.A.; Goergen, J.; Cook, J.B.; Johnson, N.; Mensah-Zoe, B.;
Simmons, J.O.; Yin, W.; Cook, J.M.; et al. Dopamine and Benzodiazepine-Dependent Mechanisms
Regulate the EtOH-Enhanced Locomotor Stimulation in the GABA A α 1 Subunit Null Mutant Mice.
Neuropsychopharmacology 2007, 32, 137–152.
148. Mihalek, R.M.; Bowers, B.J.; Wehner, J.M.; Kralic, J.E.; Vandoren, M.J.; Morrow, A.L.; Homanics, G.E.
GABAA-Receptor δ Subunit Knockout Mice Have Multiple Defects in Behavioral Responses to Ethanol.
Alcohol. Clin. Exp. Res. 2001, 25, 1708–1718.
149. Liu, J.; Yang, A.R.; Kelly, T.; Puche, A.; Esoga, C.; Elnabawi, A.; Merchenthaler, I.; Sieghart, W.; June, H.L.;
Aurelian, L. Binge alcohol drinking is associated with GABAA α2-regulated Toll-like receptor 4 (TLR4)
expression in the central amygdala. Proc. Natl. Acad. Sci. USA 2011, 108, 4465–4470.
150. Rewal, M.; Jurd, R.; Gill, T.M.; He, D.; Ron, D.; Janak, P.H. α4-Containing GABAA Receptors in the Nucleus
Accumbens Mediate Moderate Intake of Alcohol. J. Neurosci. 2009, 29, 543–549.
151. Edenberg, H.J.; Dick, D.M.; Xuei, X.; Tian, H.; Almasy, L.; Bauer, L.O.; Crowe, R.R.; Goate, A.; Hesselbrock, V.;
Jones, K.; et al. Variations in GABRA2, encoding the alpha 2 subunit of the GABA(A) receptor, are associated
with alcohol dependence and with brain oscillations. Am. J. Hum. Genet. 2004, 74, 705–714.
152. Bierut, L.J.; Agrawal, A.; Bucholz, K.K.; Doheny, K.F.; Laurie, C.; Pugh, E.; Fisher, S.; Fox, L.; Howells, W.;
Bertelsen, S.; et al. A genome-wide association study of alcohol dependence. Proc. Natl. Acad. Sci. USA 2010,
107, 5082–5087.
153. Lewohl, J.M.; Nunez, Y.O.; Dodd, P.R.; Tiwari, G.R.; Harris, R.A.; Mayfield, R.D. Up-Regulation of MicroRNAs
in Brain of Human Alcoholics. Alcohol. Clin. Exp. Res. 2011, 35, 1928–1937.
154. Sinirlioglu, Z.A.; Coskunpinar, E.; Akbas, F. miRNA and mRNA expression profiling in rat brain following
alcohol dependence and withdrawal. Cell. Mol. Biol. 2017, 63, 49–56.
155. Kumar, S.; Kralic, J.E.; O’Buckley, T.K.; Grobin, A.C.; Morrow, A.L. Chronic ethanol consumption enhances
internalization of α1 subunit-containing GABAA receptors in cerebral cortex. J. Neurochem. 2003, 86, 700–708.
156. Kumar, S.; Suryanarayanan, A.; Boyd, K.N.; Comerford, C.E.; Lai, M.A.; Ren, Q.; Morrow, A.L. Ethanol
Reduces GABAA α1 Subunit Receptor Surface Expression by a Protein Kinase Cγ-Dependent Mechanism in
Cultured Cerebral Cortical Neurons. Mol. Pharmacol. 2010, 77, 793–803.
157. Moss, S.J.; Doherty, C.A.; Huganir, R.L. Identification of the cAMP-dependent protein kinase and protein
kinase C phosphorylation sites within the major intracellular domains of the beta 1, gamma 2S, and gamma
2L subunits of the gamma-aminobutyric acid type A receptor. J. Biol. Chem. 1992, 267, 14470–14476.
158. McDonald, B.J.; Moss, S.J. Differential phosphorylation of intracellular domains of gamma-aminobutyric
acid type A receptor subunits by calcium/calmodulin type 2-dependent protein kinase and cGMP-dependent
protein kinase. J. Biol. Chem. 1994, 269, 18111–18117.
159. Parakala, M.L.; Zhang, Y.; Modgil, A.; Chadchankar, J.; Vien, T.N.; Ackley, M.A.; Doherty, J.J.; Davies, P.A.;
Moss, S.J. Metabotropic, but not allosteric, effects of neurosteroids on GABAergic inhibition depend on the
phosphorylation of GABAA receptors. J. Biol. Chem. 2019, 294, 12220–12230.
160. Kumar, S.; Lane, B.M.; Morrow, A.L. Differential Effects of Systemic Ethanol Administration on Protein
Kinase Cε, γ, and β Isoform Expression, Membrane Translocation, and Target Phosphorylation: Reversal by
Chronic Ethanol Exposure. J. Pharmacol. Exp. Ther. 2006, 319, 1366–1375.
161. Mcdonald, B.J.; Moss, S.J. Conserved phosphorylation of the intracellular domains of GABAA receptorβ2
and β3 subunits by cAMP-dependent protein kinase, cGMP-dependent protein kinase, protein kinase C and
Ca2+/calmodulin type II-dependent protein kinase. Neuropharmacology 1997, 36, 1377–1385.
162. Oh, S.; Jang, C.-G.; Ma, T.; Ho, I.K. Activation of protein kinase C by phorbol dibutyrate modulates GABAA
receptor binding in rat brain slices. Brain Res. 1999, 850, 158–165.
163. Qi, Z.-H.; Song, M.; Wallace, M.; Wang, D.; Newton, P.; McMahon, T.; Chou, W.-H.; Zhang, C.; Shokat, K.;
Messing, R.O. Protein Kinase Cε Regulates γ-Aminobutyrate Type A Receptor Sensitivity to Ethanol and
Benzodiazepines through Phosphorylation of γ2 Subunits. J. Biol. Chem. 2007, 282, 33052–33063.
164. Agrawal, A.; Pergadia, M.L.; Saccone, S.F.; Hinrichs, A.L.; Lessov-Schlaggar, C.N.; Saccone, N.L.; Neuman, R.J.;
Breslau, N.; Johnson, E.O.; Hatsukami, R.; et al. Gamma-aminobutyric acid receptor genes and nicotine
dependence: Evidence for association from a case–control study. Addiction 2008, 103, 1027–1038.
165. Balan, I.; Warnock, K.T.; Puche, A.C.; Gondre-Lewis, M.C.; June, H.; Aurelian, L. The GABAA Receptor
α2 Subunit Activates a Neuronal TLR4 Signal in the Ventral Tegmental Area that Regulates Alcohol and
Nicotine Abuse. Brain Sci. 2018, 8, 72.
166. Dixon, C.I.; Morris, H.V.; Breen, G.; Desrivières, S.; Jugurnauth, S.; Steiner, R.C.; Vallada, H.; Guindalini, C.;
Laranjeira, R.; Messas, G.; et al. Cocaine effects on mouse incentive-learning and human addiction are linked
to α2 subunit-containing GABAA receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 2289–2294.
167. Chen, Q.; Lee, T.H.; Wetsel, W.C.; Sun, Q.; Liu, Y.; Davidson, C.; Xiong, X.; Ellinwood, E.H.; Zhang, X.
Reversal of cocaine sensitization-associated changes in GAD67 and GABAA receptor α2 subunit expression,
and PKC ζ activity. Biochem. Biophys. Res. Commun. 2007, 356, 733–738.
168. Enoch, M.-A.; Zhou, Z.; Kimura, M.; Mash, D.C.; Yuan, Q.; Goldman, D. GABAergic Gene Expression in
Postmortem Hippocampus from Alcoholics and Cocaine Addicts; Corresponding Findings in Alcohol-Naïve
P and NP Rats. PLoS ONE 2012, 7, e29369.
169. Wearne, T.A.; Parker, L.M.; Franklin, J.L.; Goodchild, A.K.; Cornish, J.L. GABAergic mRNA expression
is upregulated in the prefrontal cortex of rats sensitized to methamphetamine. Behav. Brain Res. 2016,
297, 224–230.
170. Zhang, X.; Lee, T.H.; Xiong, X.; Chen, Q.; Davidson, C.; Wetsel, W.C.; Ellinwood, E.H. Methamphetamine
induces long-term changes in GABAA receptor α2 subunit and GAD67 expression. Biochem. Biophys.
Res. Commun. 2006, 351, 300–305.
171. Ammon-Treiber, S.; Höllt, V. Morphine-induced Changes of Gene Expression in the Brain. Addict. Biol. 2005,
10, 81–89.
172. Spijker, S.; Houtzager, S.W.J.; De Gunst, M.C.M.; De Boer, W.P.H.; Schoffelmeer, A.N.M.; Smit, A.B. Morphine
exposure and abstinence define specific stages of gene expression in the rat nucleus accumbens. FASEB J.
2004, 18, 848–850.
173. Heikkilä, A.T.; Echenko, O.; Uusi-Oukari, M.; Sinkkonen, S.T.; Korpi, E.R. Morphine withdrawal increases
expression of GABA(A) receptor epsilon subunit mRNA in locus coeruleus neurons. Neuroreport 2001,
12, 2981–2985.
174. Pradhan, A.A.; Tipton, A.F.; Zhang, H.; Akbari, A.; Pandey, S.C. Effect of Histone Deacetylase Inhibitor
on Ethanol Withdrawal-Induced Hyperalgesia in Rats. Int. J. Neuropsychopharmacol. 2019, 22, 523–527.
175. Tapocik, J.D.; Solomon, M.; Flanigan, M.E.; Meinhardt, M.; Barbier, E.; Schank, J.R.; Schwandt, M.;
Sommer, W.H.; Heilig, M. Coordinated dysregulation of mRNAs and microRNAs in the rat medial prefrontal
cortex following a history of alcohol dependence. Pharm. J. 2013, 13, 286–296.
176. Vithlani, M.; Hines, R.M.; Zhong, P.; Terunuma, M.; Hines, D.J.; Revilla-Sanchez, R.; Jurd, R.; Haydon, P.;
Rios, M.; Brandon, N.; et al. The ability of BDNF to modify neurogenesis and depressive-like behaviors
is dependent upon phosphorylation of tyrosine residues 365/367 in the GABA(A)-receptor γ2 subunit.
J. Neurosci. 2013, 33, 15567–15577.
177. Staley, K. Molecular mechanisms of epilepsy. Nat. Neurosci. 2015, 18, 367–372.
(翻訳&注釈:ベンゾジアゼピン情報センター 管理人)
著者:Jeffrey S Barker, Rochelle M Hines
