論文:ベンゾジアゼピン長期使用による耐性形成メカニズム:サブタイプ選択的GABAA受容体作動薬の未来は?
著者:
Christiaan H. Vinkers and Berend Olivier
投稿:2012 Mar 29
Abstract
何十年にもわたる基礎研究と臨床研究にもかかわらず、ベンゾジアゼピンが時間の経過とともにその有効性を失う(すなわち耐性がつく)傾向があることについて、我々の理解は控えめに言っても不完全である。ベンゾジアゼピンの鎮静作用と抗けいれん作用に対する耐性は比較的急速に形成される一方で、抗不安作用と健忘作用に対してはおそらく耐性がつかない。この事実を踏まえて、我々は以下に示す5つのトピックを中心に、ベンゾジアゼピン耐性の根底にある神経適応メカニズムについて、現状最新のエビデンスをレビューすることにした。(i)GABAA受容体(サブユニット発現および受容体カップリング)の変化(ii)転写および神経栄養因子に起因する細胞内変化(iii)イオンチャネル型グルタミン酸受容体(iv)その他の神経伝達物質(セロトニン、ドーパミン、およびアセチルコリン系)(v)神経ステロイド系。
さまざまな研究において、その結果が非常にばらつきが多いことから、ベンゾジアゼピンの効果ごとに異なる(そして同時に起きる)耐性メカニズムがあり、そのメカニズムは活性化されたGABAA受容体サブタイプに依存するようである。ベンゾジアゼピン部位に作用するαサブユニット選択的化合物(訳注:例として非ベンゾジアゼピン薬のザレプロン)で耐性が生じるという説得力のあるエビデンスはない。
1. Introduction
1960年代に開発されて間もなく、ベンゾジアゼピンは不安の軽減、抗けいれん作用、筋弛緩などの多くの望ましい効果を発揮し、毒性がかなり低いため非常に人気を博した[1]。しかし、鎮静、健忘症、認知障害、運動失調など、長期使用には向かない多くの副作用を生じる。ガイドラインでは一般的にベンゾジアゼピンを短期間の使用に限定しているにもかかわらず、長期使用が頻繁に発生している。慢性的なベンゾジアゼピン治療は、ベンゾジアゼピン身体依存を引き起こす可能性がある[2]。DSM-IV基準では、ベンゾジアゼピン身体依存は耐性、薬物摂取を中止したときの離脱症状など、さまざまな精神的(行動的)および身体的症状で構成される[3]。慢性的なベンゾジアゼピン治療患者は、ベンゾジアゼピンの抗けいれん、鎮静、催眠、および筋弛緩作用など、ベンゾジアゼピン作用に対する感受性が低くなる(すなわち耐性)。事実、耐性と離脱は同じ代償メカニズムから発生する2つの表現方法といえ、ベンゾジアゼピン効果がなくなった時に起きるカウンターバランスとして、離脱症状が起きるのであろう[5]。このことは、即時的なベンゾジアゼピン効果が離脱症状と正反対であり、(帯状皮質と乳頭体を含む)パペス回路でのグルコース使用変化が離脱時に観察されたという事実によって裏付けられ、離脱プロセスにおける共通回路を示唆している[6]。しかし、(通常は離脱症状の発生によって明らかになる)身体依存に必ずしも耐性がついて回るわけではなく、耐性もまた、身体依存の兆候なしに形成されることがままある[7]。
現在、何十年にもわたる基礎研究と臨床研究にもかかわらず、ベンゾジアゼピンが時間の経過とともにその有効性を失う(すなわち耐性)傾向があることについての我々の理解は、控えめに言っても不完全である。ここでは、ベンゾジアゼピン耐性の根底にある神経適応メカニズムに関する現状最新の知識を確認することとする。この論文では、ベンゾジアゼピンの精神依存性とドーパミンシステムへの影響、または乱用(大衆文化での非医療的使用)については取り上げない。それらについては[8–10]を参照いただきたい。
ベンゾジアゼピン耐性は、慢性治療における適応メカニズムであると考えられており、したがって、神経可塑性の一例と見なすことができる。古典的な(非選択的)ベンゾジアゼピンは、α1、α2、α3、またはα5サブユニットを有する抑制性GABAA受容体を調節するため、GABAA受容体の分子レベルまたは機能レベルでの耐性を解読する努力がなされてきた。一方、興奮性グルタメート系もベンゾジアゼピン耐性形成に関与していることが示唆されている[5]。慢性治療中のベンゾジアゼピン有効性低下の動的プロセスについて理解を深めることで、慢性治療中であっても有効性を保てる化合物の開発を加速できるはずである[11]。実際、さまざまなGABAA受容体サブユニットの特定機能に関するナレッジが増えることで、GABAA受容体のベンゾジアゼピン部位に作用する、より選択的な新しい薬剤が飛躍的に進歩した。アルコールは(ベンゾジアゼピンほど強力ではないが)GABAA受容体に作用するため[12]、ベンゾジアゼピン耐性とアルコール耐性を比較することは興味深いが、本論文テーマの範囲を超えており割愛する。
まず、ベンゾジアゼピン耐性形成の臨床的側面を詳しく調べる前に、GABAA受容体システムの分子基盤について説明する。次に、ベンゾジアゼピン耐性の根底にある推定分子メカニズムについて幅広く説明し、その後、古典的なベンゾジアゼピンの推定される耐性メカニズムに照らして、より選択的な新しいベンゾジアゼピンの耐性形成の問題に具体的に取り組むセクションを紹介する。既存のあるいは新規の、より選択的な薬剤を使用した有効性が持続する長期ベンゾジアゼピン治療は、患者に潜在的な利益をもたらす可能性があるので、臨床的観点から言っても耐性の理解は非常に重要である。
2. Benzodiazepines and the GABAA System
2.1. GABAA Receptors
GABAA受容体は、脳内の主な高速抑制系神経伝達物質システム構成要素である。5つの膜貫通サブユニットで構成されており、これらが集まって、さまざまなサブユニット(α1–6、β1–3、γ1–3、δ、ε、θ、およびπ)を持つリガンド開口型クロライドチャネルを形成し、GABAA受容体の不均質性につながっている[13]。GABAがGABAA受容体に結合すると、負電荷の塩化物イオンの流入を増加させ、抑制系シナプス後シグナル(IPSP)をもたらす。理論的にはサブユニットの組み合わせは膨大な数になるが、GABAA受容体の典型的なサブユニット組成は、2つのα、2つのβ、および1つのγサブユニットで構成されるものが最も一般的である[14](図1)。イン シチュ ハイブリダイゼーション(訳注:ISH法。組織や細胞において特定のDNAやmRNAの分布や量を検出する方法)および免疫組織化学研究では、GABAA受容体サブユニットが、異なる細胞局在パターンを伴うCNS分布を明確に示しており、GABAA受容体サブユニットが特殊な機能を有することを示唆している(表1)[14]。全体として、GABA作動性サブユニットは、皮質、海馬、および大脳基底核に多く発現し存在する[15]。GABA作動性サブユニットのうち、α1、β1、β2、β3、およびγ2サブユニットは脳全体に見られる。対照的に、α2、α3、α4、α5、α6、γ1、およびδサブユニットは、局所的な発現パターンを有する。α1サブユニットはβ2およびγ2サブユニットと高度に共集合しており、神経細胞体に位置している。α2またはα3サブユニットを含むGABAA受容体はそれほど多く存在せず、β3およびγ2サブユニットと共分布している。α2サブユニットは、皮質、海馬、扁桃体、視床下部に存在し、多くの場合、その発現はα1サブユニットの発現と負の相関がある。α3サブユニットの発現は、皮質、海馬、扁桃体、視床、および脳幹で最も高く、モノアミン作動性ニューロン(例、縫線核および脳幹の青斑核)および前脳のコリン作動性ニューロンでも発現する 。α5サブユニットは主に海馬で発現し、ジアゼパム感受性GABAA受容体の15〜20%を構成する[16]。細胞局在に関してであるが、形態学的に異なるGABA作動性介在ニューロンは、αサブユニットの特別なシナプス後発現を伴いながら、介在ニューロンの種類(例:シャンデリアおよびバスケット細胞)に応じて錐体細胞の異なる部分を神経支配し、皮質および海馬錐体細胞は、その介在ニューロンから入力を受け取ることになる[17、18]。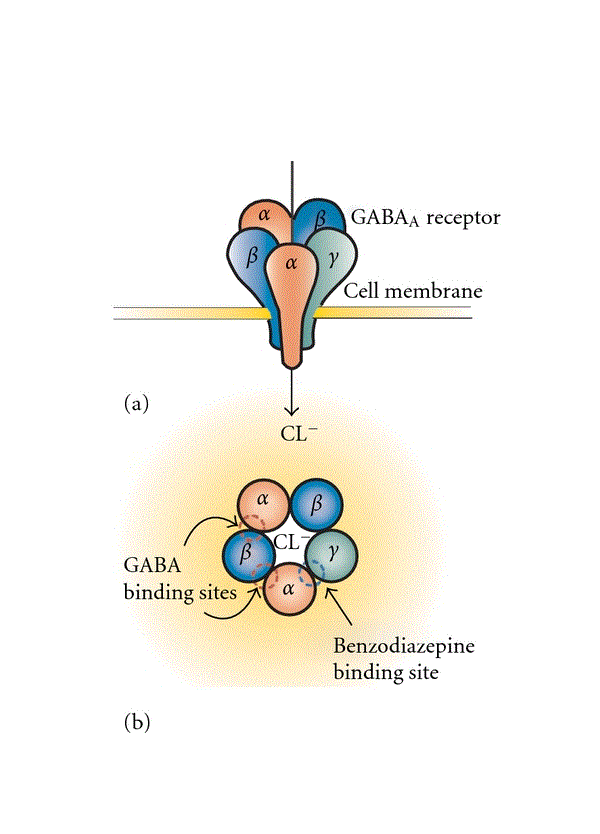
図1.GABAA受容体構造の構造表現
抑制系GABAA受容体は、リガンド開口型クロライド(Cl-)チャネルを共に形成する5つのサブユニットで構成される(a)。GABAが結合すると(GABA A受容体のαサブユニットとβサブユニットの間に)、塩化物イオンがニューロンに流れ込み、細胞膜の過分極を引き起こす(a)。古典的な非選択的ベンゾジアゼピンは、α1、α2、α3、またはα5サブユニットとγサブユニット間に結合することによってGABAの阻害作用をアロステリックに増強する(b)。GABAA受容体はサブユニットの組成に応じて大きな分子的不均質性を示すが、最も一般的なサブタイプは2α、2β、および1γサブユニットを持つ五量体となる。
表1
脳内の一般的なGABAA受容体サブタイプの局在([19]から採用)。
(表1を翻訳)
このように、GABA A受容体サブタイプはサブユニットの組成に応じて異なる機能特性を持っており、GABAシグナル伝達の複雑さに大きく関わっている[13]。付け加えると、GABAA受容体はシナプス上およびシナプス外に存在する。シナプス上の受容体は通常γサブユニットを含み、一過性の高濃度GABAを伴う高速位相抑制を仲介する[16]。対照的に、シナプス外GABAA受容体においては、GABAは(μM濃度で)より高い効力を発揮する。シナプス外GABAA受容体は通常δサブユニットを含み、α6またはα4サブユニットと優先的に集合し脱感作の動態がスローである[20]。さらに、α5サブユニットはシナプス外に局在している可能性がある[21]。シナプス外の持続的抑制作用は、あくまでベンゾジアゼピンによって変性を起こしていない場合は、脳全体における神経回路網の興奮性を調整することが示唆されている。
2.2. Benzodiazepines from a Nonspecific towards a Subunit-Specific Pharmacology
古典的なベンゾジアゼピンは、サブユニットα1、α2、α3かα5を、βひとつ、および、γ2ひとつとコンビネーションにしたGABAA受容体のベンゾジアゼピン部位に結合することで、GABA誘発IPSP(訳注:抑制性シナプス後電位)をアロステリックに調節する(図1)。正確に言うと、αサブユニットとγサブユニットの間が、GABA A受容体のベンゾジアゼピン結合部位となる。対照的に、ベンゾジアゼピンは、α4-またはα6サブユニットを含むGABAA受容体には相互作用しない。ベンゾジアゼピンに加えて、いくつかの抗けいれん薬、エタノール、バルビツール酸塩、神経ステロイド、そして、いくつかの麻酔薬など、他の薬物がGABAA受容体複合体に結合する[15]。古典的なベンゾジアゼピンが異なるαサブユニットに非選択的に結合するという事実は、抗不安剤、催眠剤、抗けいれん剤、筋弛緩剤といった薬理学的プロフィールをさらに深堀りできる可能性につながるだろう。遺伝的アプローチと薬理学的アプローチから、αサブユニットが古典的なベンゾジアゼピンのさまざまな効果に異なって寄与するという仮説を検討できる。遺伝的アプローチでは、特定のαサブユニット(α1(H101R)、α2(H101R)、α3(H126R)、およびα5(H105R))の点変異(point mutations)が、GABA感受性を変性させることなく、ベンゾジアゼピン感受性を機能的に非感受に変えてしまう、としている[22]。GABA A受容体の薬理学的研究は、さまざまなαサブユニットで異なる有効性を示す化合物の開発に注力している[13]。これらの薬物は一般的に、すべてのαサブユニット(α1、α2、α3、およびα5サブユニット)に対し等しい親和性で結合する、しかし、サブユニット1つ、あるいは2つ以上へのGABA結合能力を選択的に引き上げてしまう。こうしたストラテジーを利用して、さまざまな有効性選択的(efficacy-selective)、そして親和性選択的(affinity-selective)化合物が開発された。α1に優先的アゴニスト(ゾルピデムおよびザレプロン)やα2/3に優先的アゴニスト(TPA023、L838、417、およびSL651498)、あるいはα5に逆アゴニスト(α5IA、L-655,708、およびMRK-016)などである。表2を参照いただきたい。
表2
遺伝的および薬理学的アプローチによって、GABA作動性サブユニットの中央局在化および分布と一致する形で、GABAA受容体の異なるαサブユニットがベンゾジアゼピン効果を媒介することを明確に実証できた。具体的には、α1を含むGABA A受容体は、古典的なベンゾジアゼピンの鎮静作用、健忘作用、および抗けいれん作用を仲介する[13、23]。対照的に、ベンゾジアゼピン投与後の筋弛緩および不安軽減作用は、主にα2サブユニット(おそらくα3も)の活性化に起因していた[24]。一方、α5サブユニットを含むGABA A受容体は、学習と記憶に関与しているようである[25、26]。本レビューに照らして、ベンゾジアゼピン乱用やその耐性形成におけるGABA作動性サブユニットの関わりを調査する研究は非常に興味深いものである。しかし残念ながら、耐性形成の研究において遺伝的、サブタイプ選択的方法論を用いる研究はほとんどない。αサブユニット点変異マウスを使用した研究では、古典的なベンゾジアゼピンであるジアゼパムの慢性投与後に、その鎮静効果の低下に対し、α1サブユニットとともにα5サブユニットが重要な役割を担っていることが示唆された[27]。この研究結果については、本論文の後半で詳しく説明しよう。サブタイプ選択的GABAARモジュレーターを使用した身体依存と乱用の背景に関する研究であれば豊富にある。自己投与研究(訳注:self-administration study。おそらくサルなどの霊長類を使った実験)において、α1を含むGABAAサブタイプでの有効性が、ベンゾジアゼピンの作用強化と離脱症状に有意に関わっていることが示された[8、28、29]。具体的には、α1サブユニットで23%の固有活性(intrinsic activity)を持っているTPA123は、ベンゾジアゼピン”のような薬物の強化と離脱症状を引き起こしたが、α1固有活性が0%であるTPA023は、GABAA受容体に完全に結合する能力があっても、強化も離脱症状も引き起こさなかった[8]。ただし、TPA023はα2およびα3の有効性が低いゆえに強化および離脱を引き起こさないのかもしれない。それを裏付ける事実として、L-838,417はα1サブタイプに対する有効性を欠いているにもかかわらず、継続的な自己投与につながっている[30]。いずれにしても、α5サブユニットは古典的なベンゾジアゼピンの乱用に直接関与していない可能性がある。というのも、α5サブユニットに親和性のない、α1が大好きな催眠剤ゾルピデムはやはり霊長類での自己投与につながったからである[30]。この発見の驚くべきことは、α5サブユニットが耐性形成に関与している可能性はあるが、薬物の強化には関与してなさそう、ということである。結論としては、ベンゾジアゼピン依存症の定義に両方とも含むとしても、身体依存(のプロセス)と乱用(のプロセス)はそれぞれを独立して扱うべきである。
2.3. GABA Metabolism
ベンゾジアゼピンはGABAの抑制効果を高め、GABAの濃度-反応曲線を左にシフトするため、シナプスにおけるGABA濃度はベンゾジアゼピンの有効性に影響を与える。GABAは、細胞内においてGABAレベルを維持し、独立した2つのアイソフォーム(GAD65とGAD67)に存在する酵素である、グルタミン酸デカルボキシラーゼ(GAD)によってグルタミン酸から変換される。神経体においてGAD67が局在するのとは対照的にGAD65は主に軸索終末で発現している。このことは、シナプス神経伝達というGAD65の役割と、GABAの合成というGAD67の役割を、それぞれ示唆している[32]。シナプスGABAは、GABAトランスポーター(GAT)によって裂け目から除去され、シナプス前軸索終末に移動する。これまでに、4つのGATサブタイプが見つかっており、高度に発現されたGAT1とGAT4が最も広く分布している[33]。
3. The Development of Benzodiazepine Tolerance
長期暴露後にベンゾジアゼピン耐性形成されるその根底にあるメカニズムを調べる前に、エビデンスを検討し、エビデンスが臨床的に使用できるかを判断することは重要である。一般的に、ベンゾジアゼピンが不安や入眠潜時の軽減、けいれん予防に非常に効果的であることは疑いの余地がない。最終的に発生する耐性は、ベンゾジアゼピン効果ごとに異なる速度で異なる割合で発生するようである[34]。前臨床試験では、催眠鎮静作用への耐性がかなり急速に起こり、抗けいれん作用への耐性がそれに続き、抗不安作用への耐性は起きないか部分的であることが示されている([34– 36]を参照)。これらの前臨床試験はすでに広範囲にわたってレビューされているし、ベンゾジアゼピン耐性に関する新しい前臨床試験は、私たちの知る限りここ数年あまりないため、この論文において耐性形成に関するすべての前臨床データを再現することはできない。一般的に、前臨床試験は臨床における多様性と一致するものだが、ほとんどの前臨床試験において、耐性は用量、投与間隔、または薬剤の血漿レベルや半減期に直接関係していない。ここでは、各ベンゾジアゼピンの耐性形成(の割合)の臨床的エビデンスにのみ焦点を当てたいところだが、臨床試験が不足しているか、あるいは決定的でない場合は前臨床試験も含めることとする。
3.1. Clinical Studies on Sedative and Hypnotic Tolerance
低用量のベンゾジアゼピン身体依存症の被験者を対象とした研究において、レム睡眠抑制が依然として発生しているものの、薬の半減期とは無関係に催眠作用が失われることが示された[37]。また、他の研究では、慢性使用者は、ベンゾジアゼピンを急性投与しても鎮静または運動障害の増加を示さなかった[38–40]。さらに、ベンゾジアゼピン誘発性の反応速度低下に対する耐性は、アルプラゾラム治療の10日後には現れた[41]。短時間作用型ベンゾジアゼピンであるトリアゾラムの経口投与は、最初は睡眠導入と持続の両方を改善したが、トリアゾラム2週間使用後は、睡眠潜時と覚醒回数はベースライン値に戻ってしまった[42]。重要なことであるが、短時間作用型ベンゾジアゼピンであるトリアゾラムとミダゾラムを7日間使用後、早期覚醒が著しく悪化したのに[43]、トリアゾラムの耐性形成を示さなかった、矛盾する研究結果もまた存在する[44、45]。慢性不眠症の被験者7〜8人に26泊または54泊させ、長時間作用型ベンゾジアゼピンであるテマゼパム(15または30 mg)を使用してもらった別の研究では、テマゼパム長期投与による薬剤耐性の発生は見られなかった[46]。半減期が比較的長いフルラゼパムは、長期間使用すると日中の眠気が減少するが、中期間と長期間(4週間以上)使用において睡眠導入と持続に効果的であり続けた[47]。このように、ほとんどの研究において、鎮静効果に対する耐性はすぐに出現するのであるが、これは半減期が短いベンゾジアゼピンで顕著であるように思われる。つまり、耐性はベンゾジアゼピンの半減期によるところがありそうだ。しかしながら、短時間作用型薬物トリアゾラムではすぐに耐性がついたが、おなじく短時間作用型のミダゾラムとゾルピデムでは、ほんのわずかにしかヒト被験者の耐性が現れなかったため、この説(訳注:半減期が短いベンゾジアゼピンで耐性がつきやすいという説)は過度に一般化された認識だといえる[48]。これらの研究の限界は曝露期間が短いことにある。問題のもう1つは、長期使用において睡眠の改善がみられるという確たるエビデンスがないことである[49]。しかしこれは、耐性がついたから、というだけではなく、単に有効性の問題かもしれない。その裏付けとして、ヒト被験者では、ベンゾジアゼピンを中止しても、最大52週間までベンゾジアゼピンを服用し続けたグループと比較して、睡眠の質を低下させることはなかったし[50]、むしろ睡眠の質と徐波睡眠すらも向上させている[51]。
3.2. Clinical Studies on Anticonvulsant Tolerance
てんかんへのベンゾジアゼピン長期使用は、耐性がつくため制限されている[52]。クロバザムまたはクロナゼパムで治療されたてんかん患者の30〜50%が数か月で耐性形成される[56]。これは前臨床試験の結果に一致する[53–55]。したがって、ベンゾジアゼピンは、急性てんかん発作、あるいはてんかん重積状態でのみ、処方される。ただし、場合によっては頓服が示されることがある。頓服で耐性形成される確率が低下するだろう[57]。α1-優先化合物CL218、572でげっ歯類を慢性治療したところ、ピクロトキシン誘発性発作の喪失をもたらした[58]。古典的なベンゾジアゼピンとは対照的に、ブレタゼニルなどの部分的なGABA A受容体PAMは、いくつかの前臨床試験で抗けいれん薬耐性を示さなかった[54、59、60]。結局、私たちの知る限り、ベンゾジアゼピンは抗けいれん作用について(継続した)試験がされておらず、てんかん患者において、耐性誘発効果に関する確固たる結論には至っていない。
3.3. Clinical Studies on Amnesic Tolerance
ほとんどの研究で、慢性治療された被験者にベンゾジアゼピンを急性投与しても、短期記憶障害がみられることがわかっている[38、39、61]。また、アルプラゾラムの記憶障害効果に対する耐性は、10日間の急性治療中には見られない[41]。しかし、アルプラゾラム慢性使用後に、急性健忘症への耐性が示された、とする研究もある[40]。主な懸念として、ベンゾジアゼピン使用に関連する記憶喪失が、治療を中止した後でも持続する可能性があることだが[62、63]、ベンゾジアゼピン中止後に、情報処理の速度と精度の向上、反応時間と作業記憶の改善といった認知機能の改善が示された研究もある[50、64–66]。ベンゾジアゼピン誘発性認知障害に対する耐性が存在するという臨床データはない。
3.4. Clinical Studies on Anxiolytic Tolerance
仮に抗不安作用と催眠作用の両方とも耐性がつくとしたら、抗不安作用に対する耐性は催眠作用に対する耐性に比べてゆっくりと形成されるようである。パニック障害患者では、8週間のアルプラゾラム治療後に抗不安薬耐性も投与量増加も観察されずに有効性が継続している[67]。すでにアルプラゾラム慢性治療中のパニック障害患者を対象とした別の研究でも同様のことが確認されている。この研究では、アルプラゾラム未治療の患者と比較して、コルチゾールの応答性や抗不安薬の有効性に違いは見られなかったし[40]、疾患の重症度も関連性は特に見られない。別の二重盲検試験では、180人の慢性不安患者にジアゼパム(15〜40 mg /日)を投与し、長期のジアゼパム治療(6〜22週間)を行ったが、ジアゼパムの抗不安作用に対する耐性が生じなかった[68]。さらに、追随する研究はすべて、パニック障害[69–72]、全般性不安障害[73]、および社会恐怖症[74–76]に対して継続的な抗不安作用を示していた。ベンゾジアゼピンの長期使用後の抗不安効果の低下を明確に確認できないながらも、記憶障害、事故リスク、股関節骨折、離脱症状などその他のデメリットがあることから、不安症状に対しベンゾジアゼピンを慢性使用をすることは止めた方が良いだろう[7 、77]。結論としては、既存の文献からは、ヒトでのベンゾジアゼピンの慢性使用後に抗不安薬の有効性が低下するという確固たるエビデンスはない。
3.5. Clinical Studies on Drug Reinforcement Tolerance
ベンゾジアゼピンの薬物強化効果に対する耐性については、LicataとRowlettによってすでに議論されている[9]。彼らによると、ベンゾジアゼピンの薬物強化効果に対する耐性はありそうもないと結論付けている。この結論は、ミダゾラムとゾルピデムを継続利用可能にした環境で、非ヒト霊長類が自己注射を続け、同時に身体依存も示した研究によって裏付けられている[78、79]。ヒトの場合、薬物強化に対する耐性は、耐性と依存の悪循環サイクルを維持するための用量増加につながる可能性がある。臨床では、ほとんどの患者は用量を増やさず、薬物強化耐性が出現していない[80]。
3.6. Conclusion
結論として、耐性は、ベンゾジアゼピンの鎮静作用、催眠作用、および抗けいれん作用に対して比較的急速に形成される。 抗不安作用および健忘作用に対する耐性は、おそらくまったく現れない。ベンゾジアゼピン慢性使用後に減量が困難になるケースは、耐性形成ではなく(離脱症状が出現する)身体依存に起因していると思われる。
耐性形成の発散率とさまざまな完全性により、(i)ベンゾジアゼピン効果に応じて異なる耐性メカニズムが存在するか、(ii)メカニズムは同じであるが、GABAA受容体サブタイプのサブユニット組成と脳領域を中心とした多様性展開するもの、と推測される。しかしエビデンスからは、ベンゾジアゼピンがすべての副作用に対して堅牢で再現性のある耐性を生じると結論付けることは困難である。ベンゾジアゼピン耐性がすべての臨床効果に対して均一なプロセスではないし、利用可能ベンゾジアゼピンすべてに適用されるわけでもない。耐性を生じるかどうかを予測する因子もまた不明である。残念ながら、多くの研究がベンゾジアゼピン身体依存や乱用について扱っているが、耐性について具体的に調査する研究がない。
4. Mechanisms Underlying Tolerance
4.1. General
長期ベンゾジアゼピン治療の分子効果に関するここ数十年の研究で、わたし達の耐性についての理解は確実に進歩している。このテーマに関する優れた論文がすでにいくつも公開されている[5、11、34、77]。一般的な仮説は、ベンゾジアゼピン慢性使用が中枢神経系の適応変化につながる、というものである。GABA A受容体自体の適応変化、細胞内メカニズム、グルタミン酸作動性システムなど他の神経伝達物質システムの変化、によって、GABAA受容体がベンゾジアゼピンの継続的な急性効果に対する反応性が低下する可能性がある。適応プロセスはおそらく非常に重要な役割を果たすが、耐性形成はすべての作用について均一ではなく、前臨床と臨床とで違いが存在することを認識しておくこともまた、重要である。したがって、1つではなく複数の適応メカニズムが共存する可能性があるため、ベンゾジアゼピン耐性の研究は複雑になっている。さらに、これらの適応変化は、1つまたは複数の特定の脳領域に限定される可能性がある。このことが、耐性の根底にある先験的な統一メカニズムを1つに特定することを非常に難しいものにしている。その裏付けとなる研究がある。2-デオキシグルコース定量的オートラジオグラフィーを使用したラットにジアゼパム慢性投与すると、ジアゼパム誘発性のグルコース利用低下に対する不均一な耐性が、脳で発生した[6]。急性ジアゼパム投与は脳全体のグルコース利用低下をもたらしたが、ジアゼパム治療の3日間で、感覚処理に関連する脳部分(頭頂葉、聴覚野、蝸牛核)に耐性をもたらし、そしてそれは鎮静作用の低下と相関することが示された。28日間のジアゼパム治療後、脳グルコースに対するジアゼパムの抑制効果に対する耐性が乳頭体、小脳脚下核、および尾状核でも発生し、前頭皮質の変化が無視できないものになった。非常に興味深いのは、ベンゾジアゼピン抗不安作用が持続するのと一致して、扁桃核も経時的に鈍化を示さなかったことである。
ベンゾジアゼピン耐性の根底にある特定のメカニズムを詳しく調べる前に、おそらく、薬物動態学的要因は耐性形成に主な役割を果たしていない、ということを留意すべきである[81]。その裏付けとして、急性ジアゼパム投与後の血漿レベルは、鎮静および健忘耐性が観察されたとしても、アルプラゾラム慢性治療患者とアルプラゾラムを投与されていないパニック障害患者の間で差異はなかった[40]。ベンゾジアゼピン慢性治療後の細胞および神経機能の適応変化を媒介する最も有力な候補となるのは、GABAA受容体である。したがって最初に、ベンゾジアゼピン慢性曝露後のGABAシステムの変化(GABAA受容体カップリングとGABA受容体発現)を裏付けるエビデンスについて説明する。
4.2. GABAA System Hypotheses
4.2.1. Mechanism 1: GABAA Receptor Uncoupling
ベンゾジアゼピン機能喪失を説明するものとして、そのひとつにGABAA受容体のアロステリックなカップリングの喪失がある。GABA A受容体には、2つのGABA結合部位と1つのベンゾジアゼピン結合部位があり、ベンゾジアゼピンのベンゾジアゼピン結合部位への結合は、GABAのGABA結合部位への結合を強化する(図1)。ベンゾジアゼピンが結合すると、GABA A受容体の形態を変化させ、GABAに結合する能力を高め、チャネル開口頻度を高め、塩化物の流入を増やし、その結果過分極を引き起こすため、ベンゾジアゼピンはポジティブアロステリックモジュレーター(PAM)と呼ばれる。ベンゾジアゼピンは、GABA A受容体でGABA誘導性IPSPsを増強するが、その能力低下がGABA A受容体の脱共役と定義される。耐性に関して、ベンゾジアゼピン慢性治療はベンゾジアゼピンの薬理学的なGABA応答増強効果に対し影響を及ぼす、という仮説が立てられている(つまり、耐性は脱共役につながる)。カップリングの低下は、GABA A受容体サブユニット組成の変化、GABA A受容体自体の変化(リン酸化)またはそのセカンドメッセンジャーリガンド、あるいはGABAA受容体の形態変化の結果として発生していると思われる。受容体脱共役仮説は、サブユニット発現とリガンド結合に変化がないとしているにもかかわらず、GABAA受容体とさまざまなサブユニットの機能に関する知識を駆使しているところが魅力的だ。しかしながら、脱共役プロセスは、神経ステロイドやバルビツール酸塩など、さまざまな調節部位で作用する、また別のGABA A受容体モジュレーターへの曝露によっても誘発されるため、特殊なプロセスである[82]。
1984年には電気生理学的研究により、ベンゾジアゼピン結合部位の密度や親和性を大きく変化させることなくベンゾジアゼピン結合のGABA増強が50%減少したことがわかり、アロステリックカップリングの役割が示された[83]。最近では、GABA A受容体を発現するトランスフェクト細胞株を使用した慢性暴露実験で、アロステリックカップリングの低下の兆候が見られた[84-94]。カップリングの違いの根底にあるメカニズムは、まだよくわかっていない。GABA A受容体の組み立てプロセスが変更されているとしたら、サブユニットの置換や受容体の発現変化で、GABA受容体の組成も変更されている可能性がある。このように、GABA A受容体は、カップリングが低下するためにベンゾジアゼピン感受性が低下しているのかもしれない。私たちの知る限り、慢性暴露後のGABAA受容体サブユニット組成を直接調査した研究は存在しない。カップリングに影響を与えるメカニズムとして、GABAA受容体のリン酸化がある。GABA A受容体は、さまざまなプロテインキナーゼによってリン酸化され、ホスファターゼによって脱リン酸化される[95]。GABA A受容体のリン酸化の動的な機能変化は、チャネル開口部の変化とともに、抑制性シナプス強度に直接影響を与える可能性がある(または受容体の輸送に間接的に影響を及ぼす)。しかしながら、ニューロンのGABAA受容体機能に対するリン酸化の影響は複雑で、GABAA受容体の細胞内ループ内にまだ重要なキーが残されている。海馬ニューロンにおけるGABAA受容体IPSCの全細胞パッチクランプ記録を使用して、cAMP依存性プロテインキナーゼA(PKA)、あるいはCa2 + /リン脂質依存性プロテインキナーゼC(PKC)の活性化効果が、脳領域依存性を持って示された[96]。またPKA活性は、慢性フルラゼパム治療後の海馬錐体細胞において、GABAA受容体の変化に直接関与していることがわかった[97]。おそらく、個々の部位というよりもリン酸化パターンがより重要であり、PKAリン酸化部位への変異が、耐性に関与していないことも分かっている[90]。点変異遺伝子アプローチを使用すると、ジアゼパムの急性投与後、転写の低下は、マウスのカルシウム/カルモジュリン依存性キナーゼIIαおよびMAPキナーゼホスファターゼ-1で見られたが、α1(H101R)では見られなかった[98]。残念ながら、この研究には慢性投与の場合は含まれていなかった。
アロステリックカップリングの変化が、生体内での耐性形成に関与しているかどうかはまだ明言できない。なぜなら、ベンゾジアゼピン耐性は数日から数週間かけて徐々に形成され、このことはこの間に構造変化が起きていることを示唆しているけども、翻訳後の適応変化というものはもっと直に現れると予想されるからだ。その裏付けとして、脱共役は急速に進行するようであり、古典的なベンゾジアゼピンであるクロルジアゼポキシドは、数秒以内に単一ニューロンにおいて脱感作をもたらす[99]。また、ベンゾジアゼピン慢性治療後に観察された脱共役は、ベンゾジアゼピン拮抗薬であるフルマゼニルへの短時間曝露によって速やかに逆転する[83、86]。
4.2.2. Mechanism 2: Alterations in GABAA Receptor Subunit Expression
ベンゾジアゼピンの慢性曝露後に感受性能が落ちるメカニズムを説明する最もシンプルな仮説は、脳全体におけるGABAA受容体のダウンレギュレーションである。確かに耐性のプロセスには、少なくともある程度GABAA受容体が必要であり、GABAA受容体を発現する細胞株は、耐性の影響を受けやすい[86、87、90]。古典的な(非選択的)ベンゾジアゼピンは、α1、α2、α3、そしてα5サブユニットを含むGABA A受容体に結合するため、これらのαサブユニット(およびγ2サブユニット)で構成される受容体の発現が変化すると予想できる。もちろんこれは、細胞とGABAA受容体の解剖学的分布に依存する。すでにセクション2.1の前半で、中枢神経におけるGABA作動性サブユニットの多様で独特な分布について述べた。ベンゾジアゼピン感受性αサブユニットは、脳全体で広範に発現するが、その他のαサブユニット(α2、α3およびα5)は制限された発現パターンを示す(表1を参照)。受容体の内在化が単にGABAA受容体密度をダウンレギュレートするのなら、受容体の分布に基づいた演繹的な局所分化になるだろう。
GABA A受容体のアセンブリ、膜輸送、シナプス蓄積を制御するプロセスは複雑である([100]を参照)。簡単に言えば、GABA A受容体は、N末端のアミノ酸配列がGABA A受容体サブタイプに影響を与えながら、翻訳後数分以内に小胞体の個々のサブユニットから組み立てられる(図2)。次に、原形質膜への受容体輸送が起こり、それは多様なヘルパーGABAA受容体関連タンパク質(GABARAP、BIG2、PRIP、ゲフィリン、およびラディキシンの中で)によって促進される。最終的に(クラスリン依存性の)エンドサイトーシスが受容体の脱リン酸化後に起こり、そして分解されるかリサイクルされる(図2)。GABAシステムの長期活性化が受容体のダウンレギュレーションにつながると考える場合、これは動的なGABAA受容体のライフサイクルの複数のステップに干渉することによって確立されるといえる。ダウンレギュレーションには、サブユニットmRNA転写の減少、小胞体でのサブユニット分解(例えば、ユビキチン化によって)、GABA A受容体関連ヘルパータンパク質の発現の減少、および特定のGABAA受容体サブタイプのエンドサイトーシスの変化などがある。組換え細胞で、タンパク質合成阻害剤であるシクロヘキシミドとRNA合成阻害剤であるアクチノマイシンDが、GABA A受容体発現への慢性ジアゼパム曝露の影響を遮断した。このことは、GABAA受容体合成がある程度は重要であることを示している[87]。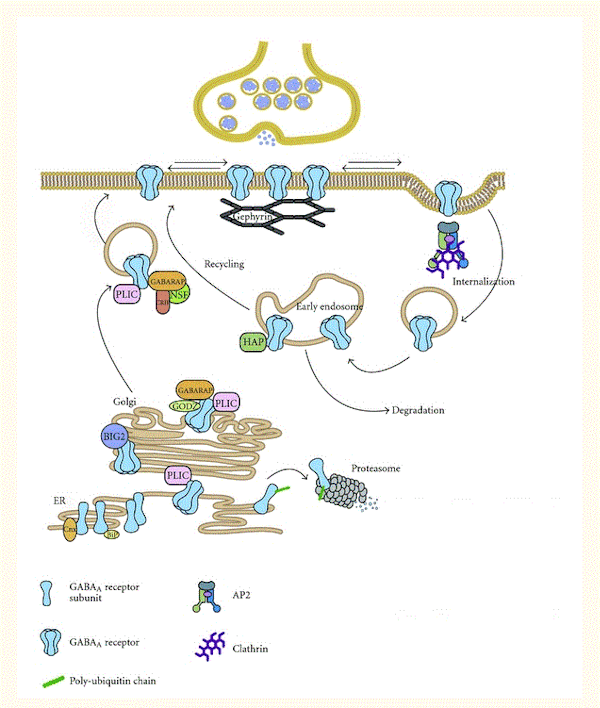
図2 GABAA受容体輸送および関連タンパク質
GABA A受容体は、シャペロンBiPとカルネキシンが品質管理を支援する小胞体(ER)の個々のサブユニットで組み立てられる。ER分解の標的となる、組み立てに使われなかったGABAA受容体サブユニットは、ユビキチン化されプロテアソームで分解される。ユビキチン様タンパク質PLICはGABAA受容体と相互作用し、それによってプロテアソーム分解の標的化を阻害する。組み立てられた五量体GABAA受容体は、ERを出てゴルジ体のグアニジン交換因子ブレフェルジンA阻害GDP / GTP交換因子2(BIG2)に結合する。ここでは、パルミトイラーゼトランスフェラーゼGODZおよびガンマアミノ酪酸受容体関連タンパク質(GABARAP)とも相互作用する。GABARAPは、NEM感受性融合(NSF)タンパク質と相互作用し、この結合により受容体複合体の細胞表面への輸送が促進される。GABA A受容体はシナプス外部位に挿入され、細胞膜に沿ってシナプスドメインの内外に拡散する。シナプスでは、GABA A受容体は足場タンパク質であるゲフィリンと相互作用し安定化される。GABA A受容体の細胞内ループとアダプチン複合体AP2のμ2サブユニットとの相互作用は、GABAA受容体の内在化にとって重要である。GABA A受容体は、クラスリンを介した経路によって初期エンドソームに送達され、そこでリソソームでの分解の標的になるか、ハンチントン関連タンパク質(HAP1)の結合時にリサイクルの標的となる。Elsevierの許可のもと[101]から転載。
これまで、さまざまなサブタイプ選択性プロファイルを持つ化合物を、さまざまな用量、さまざまな治療期間で使用し、慢性ベンゾジアゼピン治療が実際にGABA A受容体発現に影響を与えるかどうかを調べる研究が数多く行われてきた。最近の優れた研究論文では、主にラットでの実験で得られた、ベンゾジアゼピン慢性治療後のGABAA受容体サブユニットの調節に関するすべてのデータをまとめている[102]。ここではそれらの論文に記載されている綿密な作業を繰り返して確認することまではしない。サブユニットのうち、主にα、β、およびγサブユニットが調査されている。これら利用可能なエビデンスは多岐にわたり、時には矛盾しているようであるが、わたし達はmRNAとタンパク質サブユニットレベルの両方で、研究のほとんどは本質的にサブユニット発現に有意差を示していないことを確認している[102]。さらに、さまざまな脳領域においてサブユニットの発現変化に一貫性の欠如がみられる。さらに付け加えるに、ラットにおけるジアゼパム慢性治療後のGABA A受容体サブユニットmRNAレベルの差異は、慢性治療の方法によっても違いが生じる可能性がある。というのも、ジアゼパムが毎日全身注射で投与されるか、浸透圧ミニポンプで投与されるかに依存する可能性があるからである[103]。また、結合に関する研究は総じて、慢性治療後のベンゾジアゼピン結合変化を報告していない[92、93、104]。そして、GABA A受容体の発現が(mRNAレベルとタンパク質レベルの両方で)、さまざまな長期治療レジメンで一貫して確実に変化するわけでもない。したがって、ベンゾジアゼピン慢性使用後のGABA A受容体発現の一貫したダウンレギュレーションという観点からは、中枢ダウンレギュレーションのみならず、領域に特化した変化すらも、文献によって完全に支持されているわけではない。方法論の差異(例えば、治療計画、種類、投与経路、薬物)が、いくつかの矛盾生じている可能性もあるが、全体的には結果に一貫性がないように思われる。さらに、分子レベルの研究結果は行動テストと組み合わされないことが多く、行動耐性と分子変化との直接的な相関関係を考慮しないものになってしまう。ベンゾジアゼピン慢性治療後の生体内の結合と発現についての臨床研究は、それを書く人がなかなかいない。
GABA A受容体エンドサイトーシス、受容体膜挿入、細胞内輸送、およびヘルパーGABA A受容体関連タンパク質との関連するレート変化は、依然として意味を持ち、サブユニットタンパク質全体の発現に影響を与えることなく、細胞膜表面の受容体減少につながる可能性がある(e.g、[105 ])。また、慢性曝露後のシナプスの機能喪失は、GABAA受容体がGABAA受容体のクラスターから離れ、シナプス周囲またはシナプス外に局在してしまったため、という興味深い説がある(図2)[106]。少なくともアルコール研究では、抑制性シナプスにおいて可塑性の動的変化が示されている[107]。さらに、特定のサブユニットが、直接的なアップレギュレーションまたはダウンレギュレーションがない場合に、慢性治療後の耐性形成に一役買っている可能性を排除することもできない。前述のαサブユニット点変異マウスを使用すると、1日あたり15mg / kgのジアゼパム慢性治療後(8日間)であっても、急性投与されたジアゼパムはα5(H105R)マウスの自発運動活動を低下させた[27]。これは、鎮静効果を媒介するα1サブユニットが応答性を維持していることを示唆しており、古典的なベンゾジアゼピンの運動低下効果に対し耐性がつくためには、α1サブユニットとα5サブユニットの同時活性化が必要であるかもしれない。具体的には、位相シグナル伝達の増加は、α5含有GABAA受容体によって媒介されるシナプス外の持続的抑制を変化させると仮説が立つが、一方で海馬のα5特化型の結合減少がジアゼパム耐性マウスで報告されている。また、α1、α2、α3を持つ受容体とは対照的に、α5を持つGABA A受容体は、興奮性グルタミン酸作動性入力を調節できる樹状突起の基部シナプス外に位置している。ただし、α1(H101R)マウスはベンゾジアゼピン急性効果に敏感ではないため、単離されたα1サブユニット活性化との比較は不可能である。さらに、ジアゼパムの鎮静作用に対する耐性のみが報告されている。 したがって、他のベンゾジアゼピン効果に対する耐性が、他のサブユニットによって媒介される可能性はあると思われる。
4.3. Glutamate System Hypotheses
4.3.1. General
前セクションのことから、ベンゾジアゼピン慢性治療後に生じる耐性について、GABAシステムの観点から生じる代償変化だけでは、そのメカニズムの一部しか説明できないと我々は結論付けた。グルタメートは、グルタミン酸受容体に作用する興奮性神経伝達物質である。それはシナプス可塑性を調節する、GABAシステムとともに2つの対になった速効性のある神経伝達物質システムである。その証拠に、GABA作動性ニューロンとグルタミン酸作動性ニューロンの間には、密接な神経解剖学的接続が存在する[108、109]。抑制性GABAと興奮性グルタメートは、中枢神経のシナプスの少なくとも30〜50%に存在し、2つが一緒に機能することで脳の興奮バランスをとる。これら2つの相反する速効性神経伝達物質システムが微妙なバランスを形成するため、ベンゾジアゼピン慢性治療によって、GABA作動性システム活性が増加しグルタミン酸作動性伝達を混乱させることは自明のことである。ベンゾジアゼピン耐性のベースにあるのは、グルタミン酸作動性システムの感作であろうと思われる。このプロセスが、ベンゾジアゼピン中止後の離脱症状の原因であるかもしれない[5、110]。このような感作は、キンドリング実験で見られるような適応グルタミン酸作動性プロセスを彷彿とさせるが、キンドリングは断続的治療にのみ発生し、継続的な治療後では発生しないことに注意してほしい[111]。したがって、グルタミン酸作動性感作は、治療の中止時における耐性形成のみならず、離脱症状の発生にも一役買っている可能性がある。ベンゾジアゼピン離脱後のグルタミン酸作動性の変化についてはここでは説明しないが、グルタミン酸作動性システムが、不安と発作の増加を伴う離脱状態においてなんらかの役割を担っている可能性がある([5]を参照)。しかし、グルタミン酸受容体のmRNAとタンパク質の変化は離脱中に動的であり、離脱の初期段階でそのレベルは変化せず、数日後に変化が起こる[112]。その結果、離脱研究の解釈とベンゾジアゼピン耐性の理解を複雑にしている。
GABA作動性システムと同様に、グルタメートシステムは多様で複雑であり、大きくはイオノトロピック受容体タイプと代謝型受容体タイプに分けられる。イオンチャネル型グルタミン酸受容体は、グルタミン酸結合後にK +、Na +、またはCa2 +イオンの流入を増強するヘテロマーからなるリガンド作動性イオンチャネルを形成する。3つのタイプのイオンチャネル型グルタミン酸受容体がヘテロ中枢神経系で発生する:NMDA受容体(N-メチル-D-アスパラギン酸)、AMPA受容体(アルファ-アミノ-3-ヒドロキシ-5-メチル-4-イソオキサゾール-4-プロピオン酸)、およびカイニン酸受容体である(最新のレビューについては[113]を参照)。機能的なNMDA受容体は、2つの必須GluN1サブユニットと2つの調節GluN2 / 3サブユニットを持ち、シナプス可塑性に不可欠である([114]を参照)。各GluNサブユニットは、共アゴニストであるグリシンまたはD-セリン(GluN1およびGluN3サブユニットに)、およびグルタメート(GluN2サブユニットに)が結合する細胞外ループを持つ[115]。チャネルはMg2 +イオンによってブロックされているが、膜電位の変化により、Na +、Ca2 +、およびK +イオンに対して透過的になる。GluN2サブユニットは中央に分布し、NMDA受容体システムは多様になる。AMPA受容体は、4種類のサブユニット(GluR1~4)で構成されるヘテロ四量体リガンド作動性イオンチャネルであり、長期増強などの長期シナプス可塑性に不可欠である([116]を参照)。NMDA受容体と比較してAMPA受容体はグルタメート親和性が低いが、AMPA受容体にはより速い興奮誘発動態が存在する。AMPA受容体の脱感作は、イオンチャネルを閉じることを可能にするドメインインターフェースの破裂によって引き起こされることが明らかになり、シンプルでありながらエレガントな説明が可能になった[117]。カイニン酸受容体は、4つのサブユニット、GluR5、GluR6、GluR7、KA1、およびKA2で構成され、AMPAおよびNMDA受容体サブユニットに類似しており、さまざまな方法で機能的な四量体を形成する([118]を参照)。NMDAおよびAMPA受容体と比較して、カイニン酸受容体はゆっくりとした上昇および減衰特性を示す。
4.3.2. Mechanism 3: Role of Ionotropic Glutamatergic Receptors
いくつかの研究では、耐性の発生を説明するベンゾジアゼピン慢性曝露中の代償性グルタミン酸感作仮説に取り組んでいる([5,110])。
げっ歯類では、古典的なベンゾジアゼピンであるジアゼパムとクロルジアゼポキシドの鎮静作用に対する耐性形成は、NMDA受容体拮抗薬であるCPP、ジゾシルピン、MK-801、およびケタミンの同時投与によって防ぐことができた[119-121]。また、ロラゼパムの急性抗けいれん作用に対する耐性は、CPPの同時治療によって部分的に予防できた[122]。対照的に、社会的相互作用試験におけるジアゼパムの抗不安作用に対する耐性形成は、ジゾシルピン投与によって防げなかった[123]。これは、ジアゼパムの抗不安作用に対する耐性メカニズムが、鎮静作用に対する耐性メカニズムとは異なることを示唆している。NMDA NR1およびNR2BサブユニットのmRNAが、ジアゼパムに耐性がついたラットで報告されており[124、125]、NMDA受容体拮抗薬MK-801との併用使用で予防できた[126]。しかし別の研究では、フルラゼパム慢性治療後に、NMDA受容体の総量には変化がなかったが、海馬のNR2Bサブユニット減少が示された[127]。
裏付けとして、長期(急性ではない)ロラゼパム治療後、海馬でのグルタメート放出増加とNMDA誘発cGMP流出が見られたにもかかわらず、NMDA受容体の親和性あるいは密度に差がなかった[128]。まとめると、これらのデータは、NMDA依存性メカニズムがベンゾジアゼピン耐性形成に寄与していると思われる。しかし、抗不安薬耐性はNMDA受容体拮抗作用によって遮断されなかったため、NMDAシステムは特定の行動効果によって異なる働きをしているのかもしれない[123]。さらに、21日間のロラゼパム治療後の、ロラゼパムの鎮静効果に対する耐性は、グルタミン酸に対する感受性の増加ではなく減少と相関していたため、グルタミン酸感作の解釈は単純化されすぎている可能性がある([(3)H]グルタメート結合)[129]。
AMPA受容体拮抗薬GYKI52466は、ベンゾジアゼピンの長期曝露後の、ジアゼパムの鎮静作用に対する耐性形成に影響を与えなかったが[121]、AMPA受容体サブユニットの変化がが見られた[130]。具体的には、mGLuR1(皮質および扁桃体)およびmGluR2 mRNA(扁桃体)の有意な減少が、ジアゼパム慢性治療後のラットで見られた。しかし、効果は複雑で、投与経路(皮下注射か腹腔内注射)によって差異もあった。公開データの複雑さに加え、別の研究では、mEPSCが見つかり、AMPA受容体拮抗薬[(3)H] Ro48- 8587で非特異的結合が増加したにもかかわらず、フルラゼパム慢性治療後の海馬にてGluR1-3サブユニットタンパク質の変化は示されなかった [131]。GluR1ノックアウトマウスを用いた遺伝的アプローチでは、フルラゼパム準慢性治療後に、フルラゼパムの筋肉弛緩および鎮静効果に対する耐性の低下と不完全性を示した[132]。ノックアウトマウスと野生型マウスで急性フルラゼパム効果が等しかったにもかかわらず、である。
グルタミン酸作動性カイニン酸受容体については、耐性形成を調査する薬理学的または遺伝学的研究は見つけることができなかった。
まとめると、NMDA受容体の遮断がベンゾジアゼピン効果に対する耐性を防ぐ印はあるものの、汎用的で再現可能なグルタミン酸作動性成分には、一連のエビデンスからはなかなか行き着かない。しかし、分子データは多様であり時には一貫性もなく、これもまたベンゾジアゼピン慢性治療後のGABAシステム分子変化を彷彿とさせる(セクション4.2.2のとおり)。
4.4. Other Mechanisms
4.4.1. Mechanism 4: Transcriptional and Neurotrophic Factors
ベンゾジアゼピン慢性曝露に反応し下流へのシグナル伝達イベントが調整される、という仮説はもっともらしいのであるが、この分野のデータは驚くほど不足している。ベンゾジアゼピン長期使用後の、GABARAP、BIG2、PRIP、ゲフィリン、ラディキシンなど多彩なヘルパーGABAA受容体関連タンパク質の発現について思いを馳せるのは面白い(図2)。さらに、細胞内のサイクリックAMP応答配列結合タンパク質
(CREB)は、さまざまなセカンドメッセンジャーシステムで不可欠なのだが、変化する可能性もあるし、神経細胞培養においてGABA濃度が電位開口型カルシウムチャネルに影響を与えることが示されている [133]。しかしながら、ベンゾジアゼピン慢性使用誘発性のダウンストリーム細胞内変化についてはさらなるエビデンスが必要であり、今のところはまだ決定的ではない。
神経栄養タンパク質は、チロシンキナーゼ受容体(Trk)を介して、また低い親和性でp75受容体(p75NTR)を介して、神経細胞の生存、シナプスの成長、および脳全体の分化をサポートする[134]。これまでに発見されてる神経栄養因子として、脳由来神経栄養因子(BDNF)、ニューロトロフィン-3(NT-3)とニューロトロフィン-4(NT-4)、および神経成長因子(NGF)がある。いずれもシナプス高速抑制を調節する強力な因子なので、ベンゾジアゼピン慢性治療後の耐性につながる適応変化は、これら神経栄養因子によって部分的に仲介されている可能性がある。その裏付けとして、TrkB受容体の活性化を介して、BDNF(およびNT-4)がシナプス後GABAA受容体の免疫反応性を急激に低下させることがわかっている[135–139]。いっぽうで上昇が報告された研究もあり[140]、BDNF慢性治療がGABA作動性抑制を増強したと報告する研究もある[141]。この免疫反応性の低下は、GABA A受容体の発現低下によって引き起こされ、GABAA受容体アゴニストであるムシモールによるシナプス後反応の低下を伴うという仮説がある[142]。そして機構的に、GABA作動性シグナル伝達のBDNF誘発抑制は、GABA A受容体組成の変化、GABA A受容体リン酸化の増加、サブユニット合成の減少、あるいはシナプス後受容体の内在化/拡散に起因すると仮説付けられた[139]。面白いことに、こうした仮説のメカニズムはすべて本論文ですでに説明してきている。したがって、ニューロトロフィン誘発性変化は独立したメカニズムではないかもしれないが、一連のイベントにおけるプレーヤーであるのだろう。繰り返しになるが、神経栄養の発現と機能におけるベンゾジアゼピン慢性治療の影響、といった研究は私たちの知る限り存在しない。
4.4.2. Mechanism 5: Serotonin, Dopamine, Acetylcholine Systems
セロトニン、ドーパミン、およびアセチルコリン受容体システムがGABA A受容体の機能を調節するという十分なエビデンスがある[143–146](図3)。たとえば、活性化Cキナーゼの受容体(RACK-1)は、ムスカリン性アセチルコリン受容体の活性化によって媒介されるGABA A受容体のPKC依存性リン酸化を増強するし[145]、セロトニン作動性神経伝達は、やはりRACK-1とともに、GABAA受容体PKC依存性リン酸化を介してGABA作動性シグナル伝達を阻害した[144].。全体として、これらの神経伝達物質システムは、Gタンパク質共役型受容体を介して作用することで、プロテインキナーゼ(PKAおよびPKC)および足場タンパク質を活性化し、GABAA受容体のβとγ2サブユニットのリン酸化を調節する(図3)[95]。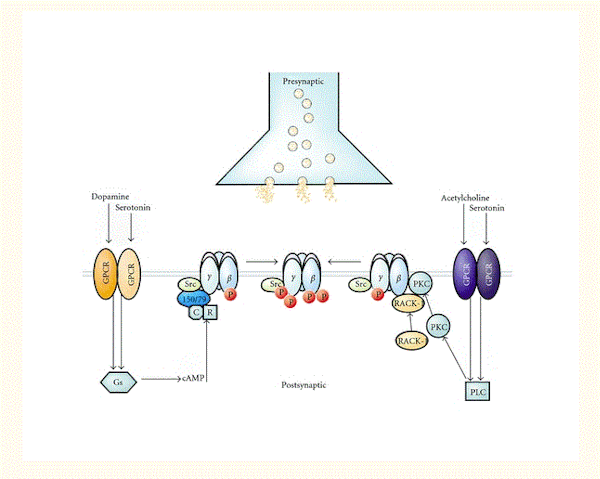
図3 (セロトニン、ドーパミン、アセチルコリン系に存在する)Gタンパク質共役型受容体(GPCR)とGABA A受容体の間の機能的クロストークは、複数のプロテインキナーゼと足場タンパク質によって促進される。GABA A受容体のβとγ2サブユニットは、ドーパミンおよびセロトニンの個々のGPCRが活性化されると、PKAおよびPKCによってリン酸化(P)される。GABAA受容体β1とβ3サブユニットのPKAリン酸化は、これらサブユニットと直接相互作用するAKAP150 / 79に依存する。AKAP150 / 79は、調節(R)および触媒(C)サブユニットで構成される不活性PKAにも結合する。さらに、PKCはβ1~3およびγ2サブユニットをリン酸化する。GPCRが活性化されると、PKCを介したリン酸化は、活性化Cキナーゼ受容体(RACK-1)とPKCのβアイソフォームが、GABA A受容体β1~3サブユニットと直接的に(ただし独立して)相互作用することで促進される。RACK-1は、これらタンパク質に関連するPKC活性を制御することで、GABAA受容体の機能的調節を促進する。GABA A受容体γ2サブユニットもSrcによってリン酸化され、このキナーゼは受容体βとγ2サブユニットに結合する。最後に、リン酸化の機能的効果は多様であり、受容体サブユニットの組成に応じて、GABAA受容体活性を阻害もするし増強もする。Elsevierの許可のもと[95]から転載。
しかしながら、ベンゾジアゼピン慢性治療におけるセロトニン、ドーパミン、およびアセチルコリンシステムの役割を調査する研究はほとんどない。健康な男性ボランティアにおける3週間のジアゼパム治療(25 mg /日)試験は、5-HT前駆体L-トリプトファンによって誘発されたプロラクチンおよび成長ホルモン反応に対する耐性を引き起こした。このとき、L-トリプトファンの鎮静効果はいちおう残ってはいた[147]。別の研究では、ジアゼパム慢性治療により、ジアゼパム耐性をもたらしただけでなく、5-HT1A受容体アゴニストである8-OH-DPATの効果がわずかに低下した[148]。対照的に、ジアゼパム慢性治療ではなく急性投与だけがラットの基底細胞外ドーパミンレベルを低下させたが、急性、慢性の両方ともストレス誘発性の皮質ドーパミンレベル上昇を逆転させることができた[149]。
4.4.3. Mechanism 6: Neurosteroids
神経ステロイドがGABAA受容体と相互作用して、強壮性(シナプス外)および位相性(シナプス上)抑制を調節する内因性アロステリックレギュレーターであるという十分で説得力のあるエビデンスがある([150、151]を参照)。また、神経ステロイド治療は急性、慢性ともに、GABA A受容体サブユニットの発現、特にシナプス外のα4およびδサブユニットを変化させる可能性がある[151]。抑制性シグナル伝達に対する神経ステロイドの可塑性誘導作用に照らして、ベンゾジアゼピンによるGABAシステムの長期的な強化は、神経ステロイドの合成や代謝の変化など、神経ステロイドシステムの変化を引き起こすが、そのような変化は古典的なベンゾジアゼピンの効果によって異なる[152]。その証拠に、卵巣摘出によってジアゼパムの抗けいれん作用に対する耐性形成を減少させた[153]。さらに、神経ステロイドのアロプレグナノロンまたはプレグネノロン(デヒドロエピアンドロステロンではない)を同時投与することによって、トリアゾラムあるいはジアゼパム慢性治療後の耐性の発生を防いだ[154]。ベンゾジアゼピン耐性への神経ステロイドの関わりが複雑である上に、GABA A受容体サブユニット組成、リン酸化メカニズム、および((余分な)シナプス)局在化などの要因が神経ステロイド活性のダイナミクスに影響している。
4.4.4. Conclusion
ベンゾジアゼピン耐性のさまざまなメカニズムに関する文献をレビューしてきて、公開データにはかなりのばらつきがあることに気づかされる。こうしたデータの不均一性は、方法論、種類、治療計画、ベンゾジアゼピンの違いによるところが大きい。具体的には、古典的なベンゾジアゼピンはすべて、α1、α2、α3、またはα5サブユニットを含むGABA A受容体に非特異的増強をもたらす、ということから、すべて同じ薬物と見なしてきた。しかしながら、生体内での薬力学的効力と薬物動態学的半減期の違いは、耐性プロセスに大きな影響を与えると言ってよい[7]。その裏付けとして、さまざまな古典的ベンゾジアゼピンによる準慢性治療を行ったところ、マウスでFG7142誘発性発作に違いが生じた。すなわちトリアゾラム、クロナゼパム、ジアゼパムはマウスの約80%で、アルプラゾラムとミダゾラムは60%、 ロラゼパムは40%で発作を生じた[155]。驚くべきことに、クロルジアゼポキシドでは同等のGABA A受容体占有率だったにもかかわらず、沈降発作を引き起こさなかった。したがって、古典的なベンゾジアゼピンがみな同じ機能だと仮定してしまうと、おそらく現在までの文献データをうまく解釈できなくなってしまう。
全体として、提示されているいずれの仮説メカニズムも、耐性形成について十分には説明できないように思われる。したがって、複数のメカニズムが(相乗的に)共存するか、まだ発見されていないメカニズムが存在するということだろう。GABAシステムの複雑で柔軟な性質と、ベンゾジアゼピン耐性に関する既存の文献の多様性を鑑みるに、ひとつの統一された耐性メカニズムでは単純すぎるのだ。ともかくも、提示されている仮説メカニズムは、神経栄養因子と神経ステロイドはGABAA受容体の組成とリン酸化状態の影響を受けるという事実からも、完全に独立しているわけではないといえる。残念ながらいまのところ薬理学的研究は、耐性がつかず長期治療に使える選択的ベンゾジアゼピンの新開発に役立つような、GABAA受容体プロファイルに関する明確な提案ができていない。
5. Tolerance to Novel Subtype-Selective Benzodiazepines
この章では、新しいGABAA受容体サブタイプ選択的化合物の耐性発現において、その役割評価を行ったエビデンスについてレビューする。サブユニット選択的ベンゾジアゼピンが開発されたことで、古典的なベンゾジアゼピンのさまざまな効果を分析することが可能となった(セクション2.2および表2を参照)。ただし、時間の経過とともに有効性が低下するプロセスは複雑で、単に1つのαサブユニットに起因するものではないと思われる。それでも、新薬が耐性がつきにくくなるものであるなら、これは臨床的観点から大いに歓迎される。新薬の有効性が継続すれば、GABAA受容体ベンゾジアゼピン部位作用薬の臨床使用が促進される。残念ながら、この新しい化合物を使用した場合の耐性形成について、本気で取り組んだ研究は多くない。私たちの研究室でのデータでは、GABAA-α2/α3選択的化合物TPA023で28日間治療されたマウスにおいて、ジアゼパムの体温降下、抗不安、鎮静といった効果に対する耐性が発生しなかった(表2)[156]。このことは、GABAA-α2/α3受容体を慢性的に活性化しても、急性ジアゼパム投入後の抗不安耐性を引き起こさないことを示している(未発表のデータ)。また、モルヒネとは対照的に、α2/ 3サブタイプGABAA受容体ポジティブアロステリックモジュレーターL838,417をラットに9日間治療したところ、鎮痛耐性は発生しなかった[157]。これらのデータから、GABAA-α2/α3サブタイプ選択薬を慢性投与しても耐性形成されない可能性があるか、あるいはジアゼパムの鎮静作用に対する耐性がつくには、GABAA-α1/GABAA-α5受容体を同時に活性化する必要があると思われる。後者の仮説を支持する事実として、ゾルピデムなどのα5サブユニットに結合しないリガンドは、耐性が生じる傾向が低下しており[158、159]、ゾルピデム(ミダゾラムではない)による慢性治療が、マウスとラットにおいて、鎮静作用と抗けいれん作用に対する耐性を引き起こさなかったという研究によっても証拠付けられている[160–162]。
耐性について直接調査してきた研究に加え、サブタイプ選択的化合物による(サブ)慢性治療後の急性離脱症状を調査した研究がある。α2、α3、およびα5GABAA受容体サブタイプ選択的有効性を持つ化合物は、マウスにおいて、インバースアゴニストFG-7142に反応して異なる発作感受性をもたらすことが示された[155]。ゾルピデムと、選択的化合物L-838,417(α2GABAA受容体、α3GABAA受容体、α5GABAA受容体の部分アゴニスト)およびSL651498(α2GABAA受容体、α3GABAA受容体の完全アゴニスト、α1GABAA受容体およびα5GABAA受容体の部分アゴニスト)による慢性治療を行っても、 FG-7142投与後の発作は生じなかった[31、155](表2)。同様に、TPA023(α2GABAA受容体、α3GABAA受容体、α5GABAA受容体の部分アゴニスト)による慢性治療でも、マウスにおいてFG-7142誘発性発作を引き起こさなかった[156]。これらの研究は耐性形成については調べていないため、かなりおおまかな結論としては、GABAA受容体を部分的あるいは選択的に調節することで、身体依存になる要因を減らせるいうことになる。こうしてみると、ゾルピデムは明らかに耐性形成引き起こさないように見えるものの、ゾルピデムでも古典的ベンゾジアゼピン慢性治療後に見られる離脱症状に匹敵するほどの離脱症状を引き起こす可能性はあり、ここは注意すべきだ[29、77]。したがって、耐性と離脱症状は、ベンゾジアゼピン依存症においてそれぞれ別個の実体であると見なせる。その証拠に、犬に対しクロラゼプ酸慢性治療を施したところ、耐性はほとんどつかなかったが、クロラゼプ酸を突然中止すると顕著な離脱症状が現れることが示された[163]。
まとめると、今のところは、α2/α3サブタイプ選択的化合物は耐性または離脱症状を引き起こさないと結論付けることができる。現在使用されているベンゾジアゼピンよりも大幅に改善されることになるが、抗不安プロファイルはまだ決定されておらず[164]、乱用の危険性は依然として存在する[8]。しかしながら、ブレタゼニルなどの非選択的部分ポジティブアロステリックモジュレーターによる慢性治療が、抗けいれん薬耐性[54、59、60]もFG-7142による発作[155]も引き起こさなかったため、解釈には慎重を要する。これらの研究は、サブタイプ選択的であっても、古典的サブタイプ選択的化合物であっても、耐性を発生させる可能性があることを示唆している。α2/α3サブタイプ選択的または弱効力が、というよりも、α1サブユニットへの弱効果が耐性形成を妨げる要因であるという仮説も成り立つ。また、α2/α3サブタイプ選択的化合物の抗不安効果はまだ確立されていない。有効性プロファイルに加え、耐性形成はGABAA受容体サブタイプに対する化合物の親和性にも依存する可能性がる。このように、耐性プロセスは、TPA023などの有効性選択性化合物(efficacy-selective compounds)とゾルピデムなどの親和性選択性化合物(affinity-selective compounds)では異なるかもしれない。ゾルピデムなどのα1-優先的親和性選択的化合物が身体的依存を引き起こし[165]、α1サブユニットで23%の有効性を有する(しかし親和性選択的ではない)化合物TPA123もまた、身体依存をもたらしたという状況証拠がある[8]。結局、現在入手可能なエビデンスを基に、耐性とサブタイプの関係性について明確な結論を引き出すことはできない。また、選択的結合(親和性affinity)と選択的活性化(有効性efficacy)とで耐性プロセスが違うと断じることも難しい。
6. Conclusion
本稿では耐性形成について、神経伝達物質システムの変化に関する、かなり一貫性のないデータをいくつかまとめてきた。具体的には、(i)GABA A受容体(サブユニット発現と受容体カップリング)、(ii)転写および神経栄養因子に起因する細胞内変化、(iii)イオンチャネル型グルタミン酸受容体、(iv)その他の神経伝達物質(セロトニン、ドーパミン、およびアセチルコリンシステム)、(v)神経ステロイドシステム、の各レベルで起こりうる変化について調べ上げてきた。多彩な研究結果から考えると、ベンゾジアゼピン効果に応じて(同時に)異なる耐性メカニズムが発生するか、あるいは、1つだけの耐性メカニズムがGABAA受容体サブタイプに応じて異なる結果をもたらすようである。耐性がつくとは、さまざまな効果に対してさまざまな速度で発生する不均一なプロセスであり、(サブタイプ選択的)ベンゾジアゼピンのプロファイルによっても差異が生じることを考えると、後者のような仮説もあり得ることだ。適応変化は、受容体サブタイプと脳領域に応じて、異なる時間スケールで発生するようだ。この仮説に沿うと、ベンゾジアゼピンの鎮静作用と抗けいれん作用に対する耐性は比較的急速に形成されるが、抗不安作用と健忘作用に対する耐性はおそらく形成されない。古典的なベンゾジアゼピンの抗不安作用が、長期治療中に低下しないのは興味深い。(サブタイプ選択的な)ベンゾジアゼピンが耐性を引き起こすには、サブタイプの選択性に加え、GABA A受容体の効力(有効性)や生体内受容体の占有率などの別の要因がキーであるかもしれない。ブレタゼニルなどのGABAA受容体αサブユニットに対し低い効力を示す部分アゴニストが、抗けいれん薬耐性を引き起こさなかったという発見がある。このことは、αサブユニット低効果の部分アゴニストが慢性的な臨床使用をしても耐性がつきにくい可能性がある。
重要な問題は、ベンゾジアゼピン耐性形成をどのように減らすことができるかということだ。興味深い提案がある。それは、個人が勝手にその頻度を決めてしまえる頓服使用ではなく、プラセボを含めたさまざまなベンゾジアゼピン投与スケジュールを作成する、というものである。これであれば継続的な臨床効果をもたらすし(もちろん適応症による)、プラセボ効果に利用できる。また、耐性形成を減らせる可能性として、サブタイプ選択的GABAA受容体PAMに関する有望な文献が現在ある。我々がレビューしたところ、α2/α3サブタイプ選択的化合物は耐性または離脱症状を引き起こさないようである。ただし、根本的なメカニズム(α1への有効性低下、それ故に全体的な有効性の低下)は不明である。また、α1-およびα5-選択的GABA作動性ポジティブアロステリックモジュレーターも耐性がつかないと言えるか現在不明だが、選択的(そしてしばしば低効力)化合物に起因した広範囲で非特異的な耐性形成はとりあえずなさそうである。
結論。ベンゾジアゼピン慢性治療後の耐性形成は複雑なプロセスであり、複数のプロセスが同時に作用し、効果と薬剤に応じたさまざまな耐性形成率となっている。ベンゾジアゼピン部位に作用するサブタイプ選択的化合物が、古典的なベンゾジアゼピンに匹敵するレベルの耐性を引き起こすという確たるエビデンスはない。もしそうなら、サブタイプ選択的化合物が臨床において耐性を引き起こす可能性は低くなるかもしれず、古典的なベンゾジアゼピンよりも臨床的に有意な改善となると思われる。
References
1. Lader M. History of benzodiazepine dependence. Journal of Substance Abuse Treatment. 1991;8(1-2):53–59.
2. Owen RT, Tyrer P. Benzodiazepine dependence. A review of the evidence. Drugs. 1983;25(4):385–398.
3. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV-TR. 2000. [Google Scholar]
4. Petursson H. The benzodiazepine withdrawal syndrome. Addiction. 1994;89(11):1455–1459.
5. Allison C, Pratt JA. Neuroadaptive processes in GABAergic and glutamatergic systems in benzodiazepine dependence. Pharmacology and Therapeutics. 2003;98(2):171–195.
6. Pratt JA, Brett RR, Laurie DJ. Benzodiazepine dependence: from neural circuits to gene expression. Pharmacology Biochemistry and Behavior. 1998;59(4):925–934.
7. Woods JH, Katz JL, Winger G. Benzodiazepines: use, abuse, and consequences. Pharmacological Reviews. 1992;44(2):151–347.
8. Ator NA, Atack JR, Hargreaves RJ, Burns HD, Dawson GR. Reducing abuse liability of GABAA/benzodiazepine ligands via selective partial agonist efficacy at α 1 and α 2/3 subtypes. Journal of Pharmacology and Experimental Therapeutics. 2010;332(1):4–16.
9. Licata SC, Rowlett JK. Abuse and dependence liability of benzodiazepine-type drugs: GABAA receptor modulation and beyond. Pharmacology Biochemistry and Behavior. 2008;90(1):74–89.
10. Tan KR, Brown M, Labouébe G, et al. Neural bases for addictive properties of benzodiazepines. Nature. 2010;463(7282):769–774.
11. Wafford KA. GABAA receptor subtypes: any clues to the mechanism of benzodiazepine dependence? Current Opinion in Pharmacology. 2005;5(1):47–52.
12. Krystal JH, Staley J, Mason G, et al. γ-aminobutyric acid type A receptors and alcoholism: intoxication, dependence, vulnerability, and treatment. Archives of General Psychiatry. 2006;63(9):957–968.
13. Rudolph U, Mohler H. GABA-based therapeutic approaches: GABAA receptor subtype functions. Current Opinion in Pharmacology. 2006;6(1):18–23.
14. McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends in Neurosciences. 1996;19(4):139–143.
15. Sieghart W. Structure and pharmacology of γ-aminobutyric acidA receptor subtypes. Pharmacological Reviews. 1995;47(2):181–234.
16. Sieghart W, Sperk G. Subunit composition, distribution and function of GABAA receptor subtypes. Current Topics in Medicinal Chemistry. 2002;2(8):795–816.
17. Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyl P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378(6552):75–78.
18. Lewis DA, Cho RY, Carter CS, et al. Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. American Journal of Psychiatry. 2008;165(12):1585–1593.
19. Mohler H, Fritschy JM, Rudolph U. A new benzodiazepine pharmacology. Journal of Pharmacology and Experimental Therapeutics. 2002;300(1):2–8.
20. Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC, Cope DW. Extrasynaptic GABAA receptors: form, pharmacology, and function. Journal of Neuroscience. 2009;29(41):12757–12763.
21. Serwanski DR, Miralles CP, Christie SB, Mehta AK, Li X, De Blas AL. Synaptic and nonsynaptic localization of GABAA receptors containing the α5 subunit in the rat brain. Journal of Comparative Neurology. 2006;499(3):458–470.
22. Rudolph U, Crestani F, Benke D, et al. Benzodiazepine actions mediated by specific γ-aminobutyric acidA receptor subtypes. Nature. 1999;401(6755):796–800.
23. McKernan RM, Rosahl TW, Reynolds DS, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nature Neuroscience. 2000;3(6):587–592.
24. Atack JR. GABAA receptor subtype-selective modulators. I. alpha2/alpha3-selective agonists as non-sedating anxiolytics. Current Topics in Medicinal Chemistry. 2010;2:331–360.
25. Collinson N, Kuenzi FM, Jarolimek W, et al. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the α5 subunit of the GABAA receptor. Journal of Neuroscience. 2002;22(13):5572–5580.
26. Dawson GR, Maubach KA, Collinson N, et al. An inverse agonist selective for α5 subunit-containing GABAA receptors enhances cognition. Journal of Pharmacology and Experimental Therapeutics. 2006;316(3):1335–1345.
27. van Rijnsoever C, Täuber M, Choulli MK, et al. Requirement of α5-GABAA receptors for the development of tolerance to the sedative action of diazepam in mice. Journal of Neuroscience. 2004;24(30):6785–6790.
28. Ator NA. Contributions of GABAA receptor subtype selectivity to abuse liability and dependence potential of pharmacological treatments for anxiety and sleep disorders. CNS Spectrums. 2005;10(1):31–39.
29. Griffiths RR, Sannerud CA, Ator NA, Brady JV. Zolpidem behavioral pharmacology in baboons: self-injection, discrimination, tolerance and withdrawal. Journal of Pharmacology and Experimental Therapeutics. 1992;260(3):1199–1208.
30. Rowlett JK, Platt DM, Lelas S, Atack JR, Dawson GR. Different GABAA receptor subtypes mediate the anxiolytic, abuse-related, and motor effects of benzodiazepine-like drugs in primates. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(3):915–920.
31. Griebel G, Perrault G, Simiand J, et al. SL651498: an anxioselective compound with functional selectivity for alpha2- and alpha3-containing gamma-aminobutyric acidAGABAA receptors. Journal of Pharmacology and Experimental Therapeutics. 2001;298(2):753–768.
32. Benes FM, Todtenkopf MS, Logiotatos P, Williams M. Glutamate decarboxylase(65)-immunoreactive terminals in cingulate and prefrontal cortices of schizophrenic and bipolar brain. Journal of Chemical Neuroanatomy. 2000;20(3-4):259–269.
33. Schousboe A, Sarup A, Larsson OM, White HS. GABA transporters as drug targets for modulation of GABAergic activity. Biochemical Pharmacology. 2004;68(8):1557–1563.
34. Bateson AN. Basic pharmacologic mechanisms involved in benzodiazepine tolerance and withdrawal. Current Pharmaceutical Design. 2002;8(1):5–21.
35. File SE. Tolerance to the behavioral actions of benzodiazepines. Neuroscience and Biobehavioral Reviews. 1985;9(1):113–121.
36. File SE. The history of benzodiazepine dependence: a review of animal studies. Neuroscience and Biobehavioral Reviews. 1990;14(2):135–146.
37. Schneider-Helmert D. Why low-dose benzodiazepine-dependent insomniacs can’t escape their sleeping pills. Acta Psychiatrica Scandinavica. 1988;78(6):706–711.
38. Lucki I, Rickels K. The behavioral effects of benzodiazepines following long-term use. Psychopharmacology Bulletin. 1986;22(2):424–433.
39. Lucki I, Rickels K, Geller AM. Chronic use of benzodiazepines and psychomotor and cognitive test performance. Psychopharmacology. 1986;88(4):426–433.
40. Cowley DS, Roy-Byrne PP, Radant A, et al. Benzodiazepine sensitivity in panic disorder: effects of chronic alprazolam treatment. Neuropsychopharmacology. 1995;12(2):147–157.
41. Allen D, Curran HV, Lader M. The effects of repeated doses of clomipramine and alprazolam on physiological, psychomotor and cognitive functions in normal subjects. European Journal of Clinical Pharmacology. 1991;40(4):355–362.
42. Kales A, Scharf MB, Kales JD. Rebound insomnia: a new clinical syndrome. Science. 1978;201(4360):1039–1041.
43. Kales A, Soldatos CR, Bixler EO, Kales JD. Early morning insomnia with rapidly eliminated benzodiazepines. Science. 1983;220(4592):95–97.
44. Pegram V, Hyde P, Linton P. Chronic use of triazolam: the effects on the sleep patterns of insomniacs. Journal of International Medical Research. 1980;8(3):224–231.
45. Roth T, Kramer M, Lutz T. Intermediate use of triazolam: a sleep laboratory study. Journal of International Medical Research. 1976;4(1):59–63.
46. Mitler MM, Carskadon MA, Phillips RL, et al. Hypnotic efficacy of temazepam: a long-term sleep laboratory evaluation. British Journal of Clinical Pharmacology. 1979;8(1):63S–68S.
47. Kales A, Kales JD. Sleep laboratory studies of hypnotic drugs: efficacy and withdrawal effects. Journal of Clinical Psychopharmacology. 1983;3(2):140–150.
48. Soldatos CR, Dikeos DG, Whitehead A. Tolerance and rebound insomnia with rapidly eliminated hypnotics: a meta-analysis of sleep laboratory studies. International Clinical Psychopharmacology. 1999;14(5):287–303.
49. Monane M, Glynn RJ, Avorn J. The impact of sedative-hypnotic use on sleep symptoms in elderly nursing home residents. Clinical Pharmacology and Therapeutics. 1996;59(1):83–92.
50. Curran HV, Collins R, Fletcher S, Kee SCY, Woods B, Iliffe S. Older adults and withdrawal from benzodiazepine hypnotics in general practice: effects on cognitive function, sleep, mood and quality of life. Psychological Medicine. 2003;33(7):1223–1237.
51. Poyares D, Guilleminault C, Ohayon MM, Tufik S. Chronic benzodiazepine usage and withdrawal in insomnia patients. Journal of Psychiatric Research. 2004;38(3):327–334.
52. Browne TR, Penry JK. Benzodiazepines in the treatment of epilepsy: a review. Epilepsia. 1973;14(3):277–310.
53. Loscher W, Rundfeldt C, Honack D, Ebert U. Long-term studies on anticonvulsant tolerance and withdrawal characteristics of benzodiazepine receptor ligands in different seizure models in mice. I. Comparison of diazepam, clonazepam, clobazam and abecarnil. Journal of Pharmacology and Experimental Therapeutics. 1996;279(2):561–572.
54. Rundfeldt C, Wlaz P, Honack D, Loscher W. Anticonvulsant tolerance and withdrawal characteristics of benzodiazepine receptor ligands in different seizure models in mice. Comparison of diazepam, Bretazenil and Abecarnil. Journal of Pharmacology and Experimental Therapeutics. 1995;275(2):693–702.
55. Gonsalves SF, Gallager DW. Time course for development of anticonvulsant tolerance and GABAergic subsensitivity after chronic diazepam. Brain Research. 1987;405(1):94–99.
56. Ko D, Rho JM, DeGiorgio CM, Sato S. Benzodiazpines. In: Engel JJ, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. Philadelphia, Pa, USA: Lippincott-Raven; 1997. pp. 1475–1489. [Google Scholar]
57. Brodie MJ. Established anticonvulsants and treatment of refractory epilepsy. The Lancet. 1990;336(8711):350–354.
58. File SE, Wilks L. Effects of acute and chronic treatment on the pro- and anti-convulsant actions of CL 218, 872, PK 8165 and PK 9084, putative ligands for the benzodiazepine receptor. Journal of Pharmacy and Pharmacology. 1985;37(4):252–256.
59. Feely M, Boyland P, Picardo A, Cox A, Gent JP. Lack of anticonvulsant tolerance with RU 32698 and Ro 17-1812. European Journal of Pharmacology. 1989;164(2):377–380.
60. Loscher W, Rundfeldt C, Honack D, Ebert U. Long-term studies on anticonvulsant tolerance and withdrawal characteristics of benzodiazepine receptor ligands in different seizure models in mice. II. The novel imidazoquinazolines NNC 14-0185 and NNC 14-0189. Journal of Pharmacology and Experimental Therapeutics. 1996;279(2):573–581.
61. Curran HV, Bond A, O’Sullivan G, et al. Memory functions, alprazolam and exposure therapy: a controlled longitudinal study of agoraphobia with panic disorder. Psychological Medicine. 1994;24(4):969–976.
62. Tata PR, Rollings J, Collins M, Pickering A, Jacobson RR. Lack of cognitive recovery following withdrawal from long-term benzodiazepine use. Psychological Medicine. 1994;24(1):203–213.
63. Barker MJ, Greenwood KM, Jackson M, Crowe SF. Persistence of cognitive effects after withdrawal from long-term benzodiazepine use: a meta-analysis. Archives of Clinical Neuropsychology. 2004;19(3):437–454.
64. Tsunoda K, Uchida H, Suzuki T, Watanabe K, Yamashima T, Kashima H. Effects of discontinuing benzodiazepine-derivative hypnotics on postural sway and cognitive functions in the elderly. International Journal of Geriatric Psychiatry. 2010;25(12):1259–1265.
65. Tonne U, Hiltunen AJ, Vikander B, et al. Neuropsychological changes during steady-state drug use, withdrawal and abstinence in primary benzodiazepine-dependent patients. Acta Psychiatrica Scandinavica. 1995;91(5):299–304.
66. Rickels K, Lucki I, Schweizer E, García-España F, Case WG. Psychomotor performance of long-term benzodiazepine users before, during, and after benzodiazepine discontinuation. Journal of Clinical Psychopharmacology. 1999;19(2):107–113.
67. Schweizer E, Rickels K, Weiss S, Zavodnick S. Maintenance drug treatment of panic disorder: I. Results of a prospective, placebo-controlled comparison of alprazolam and imipramine. Archives of General Psychiatry. 1993;50(1):51–60.
68. Rickels K, Case G, Downing RW, Winokur A. Long-term diazepam therapy and clinical outcome. The Journal of the American Medical Association. 1983;250(6):767–771.
69. Charney DS, Woods SW. Benzodiazepine treatment of panic disorder: a comparison of alprazolam and lorazepam. Journal of Clinical Psychiatry. 1989;50(11):418–423.
70. Ballenger JC. Long-term pharmacologic treatment of panic disorder. Journal of Clinical Psychiatry. 1991;52(2, supplement):18–23.
71. Burrows GD, Judd FK, Norman TR. Long-term drug treatment of panic disorder. Journal of Psychiatric Research. 1993;27(supplement 1):111–125.
72. Pollack MH, Tesar GE, Rosenbaum JF, Spier SA. Clonazepam in the treatment of panic disorder and agoraphobia: a one-year follow-up. Journal of Clinical Psychopharmacology. 1986;6(5):302–304.
73. Cohn JB, Wilcox CS. Long-term comparison of alprazolam, lorazepam and placebo in patients with an anxiety disorder. Pharmacotherapy. 1984;4(2):93–98.
74. Sutherland SM, Tupler LA, Colket JT, Davidson JRT. A 2-year follow-up of social phobia: status after a brief medication trial. Journal of Nervous and Mental Disease. 1996;184(12):731–738.
75. Davidson JRT, Ford SM, Smith RD, Potts NLS. Long-term treatment of social phobia with clonazepam. Journal of Clinical Psychiatry. 1991;52(11, supplement):16–20.
76. Otto MW, Pollack MH, Gould RA, et al. A comparison of the efficacy of clonazepam and cognitive-behavioral group therapy for the treatment of social phobia. Journal of Anxiety Disorders. 2000;14(4):345–358.
77. Griffiths RR, Weerts EM. Benzodiazepine self-administration in humans and laboratory animals—implications for problems of long-term use and abuse. Psychopharmacology. 1997;134(1):1–37.
78. Weerts EM, Griffiths RR. Zolpidem self-injection with concurrent physical dependence under conditions of long-term continuous availability in baboons. Behavioural Pharmacology. 1998;9(3):285–297.
79. Weerts EM, Kaminski BJ, Griffiths RR. Stable low-rate midazolam self-injection with concurrent physical dependence under conditions of long-term continuous availability in baboons. Psychopharmacology. 1998;135(1):70–81.
80. Soumerai SB, Simoni-Wastila L, Singer C, et al. Lack of relationship between long-term use of benzodiazepines and escalation to high dosages. Psychiatric Services. 2003;54(7):1006–1011.
81. Fernandes C, File SE, Berry D. Evidence against oppositional and pharmacokinetic mechanisms of tolerance to diazepam’s sedative effects. Brain Research. 1996;734(1-2):236–242.
82. Friedman LK, Gibbs TT, Farb DH. γ-aminobutyric acidA receptor regulation: heterologous uncoupling of modulatory site interactions induced by chronic steroid, barbiturate, benzodiazepine, or GABA treatment in culture. Brain Research. 1996;707(1):100–109.
83. Gallager DW, Lakoski JM, Gonsalves Rauch SFSL. Chronic benzodiazepine treatment decreases postsynaptic GABA sensitivity. Nature. 1984;308(5954):74–77.
84. Wong G, Lyon T, Skolnick P. Chronic exposure to benzodiazepine receptor ligands uncouples the γ—aminobutyric acid type A receptor in WSS-1 cells. Molecular Pharmacology. 1994;46(6):1056–1062.
85. Tietz EI, Chiu TH, Rosenberg HC. Regional GABA/benzodiazepine receptor/chloride channel coupling after acute and chronic benzodiazepine treatment. European Journal of Pharmacology. 1989;167(1):57–65.
86. Primus RJ, Yu J, Xu J, et al. Allosteric uncoupling after chronic benzodiazepine exposure of recombinant γ-aminobutyric acidA receptors expressed in Sf9 cells: ligand efficacy and subtype selectivity. Journal of Pharmacology and Experimental Therapeutics. 1996;276(3):882–890.
87. Pericic D, Strac DS, Jembrek MJ, Vlainic J. Allosteric uncoupling and up-regulation of benzodiazepine and GABA recognition sites following chronic diazepam treatment of HEK 293 cells stably transfected with α1β2γ2S subunits of GABAA receptors. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2007;375(3):177–187.
88. Klein RL, Harris RA. Regulation of GABAA receptor structure and function by chronic drug treatments in vivo and with stably transfected cells. Japanese Journal of Pharmacology. 1996;70(1):1–15.
89. Klein RL, Mascia MP, Harkness PC, Hadingham KL, Whiting PJ, Harris RA. Regulation of allosteric coupling and function of stably expressed γ-aminobutyric acid GABAA receptors by chronic treatment with GABAA and benzodiazepine agonists. Journal of Pharmacology and Experimental Therapeutics. 1995;274(3):1484–1492.
90. Ali NJ, Olsen RW. Chronic benzodiazepine treatment of cells expressing recombinant GABAA receptors uncouples allosteric binding: studies on possible mechanisms. Journal of Neurochemistry. 2001;79(5):1100–1108.
91. Itier V, Granger P, Perrault G, Depoortere H, Scatton B, Avenet P. Protracted treatment with diazepam reduces benzodiazepine1 receptor-mediated potentiation of gamma-aminobutyric acid-induced currents in dissociated rat hippocampal neurons. Journal of Pharmacology and Experimental Therapeutics. 1996;279(3):1092–1099.
92. Heninger C, Gallager DW. Altered γ-aminobutyric acid/benzodiazepine interaction after chronic diazepam exposure. Neuropharmacology. 1988;27(10):1073–1076.
93. Brett RR, Pratt JA. Changes in benzodiazepine-GABA receptor coupling in an accumbens-habenula circuit after chronic diazepam treatment. British Journal of Pharmacology. 1995;116(5):2375–2384.
94. Roca DJ, Rozenberg I, Farrant M, Farb DH. Chronic agonist exposure induces down-regulation and allosteric uncoupling of the γ-aminobutyric acid/benzodiazepine receptor complex. Molecular Pharmacology. 1990;37(1):37–43.
95. Kittler JT, Moss SJ. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Current Opinion in Neurobiology. 2003;13(3):341–347.
96. Poisbeau P, Cheney MC, Browning MD, Mody I. Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. Journal of Neuroscience. 1999;19(2):674–683.
97. Lilly SM, Zeng XJ, Tietz EI. Role of protein kinase A in GABAA receptor dysfunction in CA1 pyramidal cells following chronic benzodiazepine treatment. Journal of Neurochemistry. 2003;85(4):988–998.
98. Huopaniemi, L, Keist R, Randolph A, Certa U, Rudolph U. Diazepam-induced adaptive plasticity revealed by alpha1 GABAA receptor-specific expression profiling. Journal of Neurochemistry. 2004;88(5):1059–1067.
99. Mierlak D, Farb DH. Modulation of neurotransmitter receptor desensitization chlordiazepoxide stimulates fading of the GABA response. Journal of Neuroscience. 1988;8(3):814–820.
100. Jacob TC, Moss SJ, Jurd R. GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nature Reviews Neuroscience. 2008;9(5):331–343.
101. Arancibia-Cárcamo IL, Kittler JT. Regulation of GABAA receptor membrane trafficking and synaptic localization. Pharmacology and Therapeutics. 2009;123(1):17–31.
102. Uusi-Oukari M, Korpi ER. Regulation of GABAA receptor subunit expression by pharmacological agents. Pharmacological Reviews. 2010;62(1):97–135.
103. Arnot MI, Davies M, Martin IL, Bateson AN. GABAA receptor gene expression in rat cortex: differential effects of two chronic diazepam treatment regimes. Journal of Neuroscience Research. 2001;64(6):617–625.
104. Little HJ, Nutt DJ, Taylor SC. Bidirectional effects of chronic treatment with agonists and inverse agonists at the benzodiazepine receptor. Brain Research Bulletin. 1987;19(3):371–378.
105. Tehrani MH, Barnes EM., Jr. Sequestration of gamma-aminobutyric acidA receptors on clathrin-coated vesicles during chronic benzodiazepine administration in vivo . Journal of Pharmacology and Experimental Therapeutics. 1997;283(1):384–390.
106. Barnes EM., Jr. Intracellular trafficking of GABAA receptors. Life Sciences. 2000;66(12):1063–1070.
107. Liang J, Zhang N, Cagetti E, Houser CR, Olsen RW, Spigelman I. Chronic intermittent ethanol-induced switch of ethanol actions from extrasynaptic to synaptic hippocampal GABAA receptors. Journal of Neuroscience. 2006;26(6):1749–1758.
108. Herman JP, Mueller NK, Figueiredo H. Role of GABA and glutamate circuitry in hypothalamo-pituitary-adrenocortical stress integration. Annals of the New York Academy of Sciences. 2004;1018:35–45.
109. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. Journal of Neuroscience. 2007;27(43):11496–11500.
110. Stephens DN. A glutamatergic hypothesis of drug dependence: extrapolations from benzodiazepine receptor ligands. Behavioural Pharmacology. 1995;6(5-6):425–446.
111. Marley RJ, Heninger C, Hernandez TD, Gallager DW. Chronic administration of β-carboline-3-carboxylic acid methylamide by continuous intraventricular infusion increases GABAergic function. Neuropharmacology. 1991;30(3):245–251.
112. Izzo E, Auta J, Impagnatiello F, Pesold C, Guidotti A, Costa E. Glutamic acid decarboxylase and glutamate receptor changes during tolerance and dependence to benzodiazepines. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(6):3483–3488.
113. Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological Reviews. 2010;62(3):405–496.
114. Bard L, Groc L. Glutamate receptor dynamics and protein interaction: lessons from the NMDA receptor. Molecular and Cellular Neuroscience. 2011;48(4):298–307.
115. Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Current Opinion in Pharmacology. 2007;7(1):39–47.
116. Nakagawa T. The biochemistry, ultrastructure, and subunit assembly mechanism of AMPA receptors. Molecular Neurobiology. 2010;42(3):161–184.
117. Armstrong N, Jasti J, Beich-Frandsen M, Gouaux E. Measurement of conformational changes accompanying desensitization in an Ionotropic glutamate receptor. Cell. 2006;127(1):85–97.
118. Lerma J. Kainate receptor physiology. Current Opinion in Pharmacology. 2006;6(1):89–97.
119. Khanna JM, Chau A, Shah G. Effect of NMDA antagonists on rapid tolerance to benzodiazepines. Brain Research Bulletin. 1997;42(2):99–103.
120. File SE, Fernandes C. Dizocilpine prevents the development of tolerance to the sedative effects of diazepam in rats. Pharmacology Biochemistry and Behavior. 1994;47(4):823–826.
121. Steppuhn KG, Turski L. Diazepam dependence prevented by glutamate antagonists. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(14):6889–6893.
122. Koff JM, Pritchard GA, Greenblatt DJ, Miller LG. The NMDA receptor competitive antagonist CPP modulates benzodiazepine tolerance and discontinuation. Pharmacology. 1997;55(5):217–227.
123. Fernandes C, File SE. Dizocilpine does not prevent the development of tolerance to the anxiolytic effects of diazepam in rats. Brain Research. 1999;815(2):431–434.
124. Tsuda M, Shimizu N, Yajima Y, Suzuki T, Misawa M. Hypersusceptibility to DMCM-induced seizures during diazepam withdrawal in mice: evidence for upregulation of NMDA receptors. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1998;357(3):309–315.
125. Perez MF, Salmiron R, Ramirez OA. NMDA-NR1 and -NR2B subunits mRNA expression in the hippocampus of rats tolerant to diazepam. Behavioural Brain Research. 2003;144(1-2):119–124.
126. Almiron RS, Perez MF, Ramirez OA. MK-801 prevents the increased NMDA-NR1 and NR2B subunits mRNA expression observed in the hippocampus of rats tolerant to diazepam. Brain Research. 2004;1008(1):54–60.
127. Van Sickle BJ, Cox AS, Schak K, Greenfield LJ, Tietz EI. Chronic benzodiazepine administration alters hippocampal CA1 neuron excitability: NMDA receptor function and expression. Neuropharmacology. 2002;43(4):595–606.
128. Bonavita C, Ferrero A, Cereseto M, Velardez M, Rubio M, Wikinski S. Adaptive changes in the rat hippocampal glutamatergic neurotransmission are observed during long-term treatment with lorazepam. Psychopharmacology. 2003;166(2):163–167.
129. Bonavita CD, Bisagno V, Bonelli CG, Acosta GB, Rubio MC, Wikinski SI. Tolerance to the sedative effect of lorazepam correlates with a diminution in cortical release and affinity for glutamate. Neuropharmacology. 2002;42(5):619–625.
130. Allison C, Pratt JA. Differential effects of two chronic diazepam treatment regimes on withdrawal anxiety and AMPA receptor characteristics. Neuropsychopharmacology. 2006;31(3):602–619.
131. Van Sickle BJ, Tietz EI. Selective enhancement of AMPA receptor-mediated function in hippocampal CA1 neurons from chronic benzodiazepine-treated rats. Neuropharmacology. 2002;43(1):11–27.
132. Aitta-aho T, Vekovischeva OY, Neuvonen PJ, Korpi ER. Reduced benzodiazepine tolerance, but increased flumazenil-precipitated withdrawal in AMPA-receptor GluR-A subunit-deficient mice. Pharmacology Biochemistry and Behavior. 2009;92(2):283–290.
133. Lyons HR, Land MB, Gibbs TT, Farb DH. Distinct signal transduction pathways for GABA-induced GABAA receptor down-regulation and uncoupling in neuronal culture: a role for voltage-gated calcium channels. Journal of Neurochemistry. 2001;78(5):1114–1125.
134. Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nature Reviews Neuroscience. 2005;6(8):603–614.
135. Brunig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABAA receptor surface expression. European Journal of Neuroscience. 2001;13(7):1320–1328.
136. Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ. Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABAA receptor phosphorylation, activity, and cell-surface stability. Journal of Neuroscience. 2004;24(2):522–530.
137. Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. Journal of Neuroscience. 1997;17(9):2959–2966.
138. Cheng Q, Yeh HH. Brain-derived neurotrophic factor attenuates mouse cerebellar granule cell GABAA receptor-mediated responses via postsynaptic mechanisms. Journal of Physiology. 2003;548(3):711–721.
139. Henneberger C, Jüttner R, Rothe T, Grantyn R. Postsynaptic action of BDNF on GABAergic synaptic transmission in the superficial layers of the mouse superior colliculus. Journal of Neurophysiology. 2002;88(2):595–603.
140. Mizoguchi Y, Kanematsu T, Hirata M, Nabekura J. A rapid increase in the total number of cell surface functional GABAA receptors induced by brain-derived neurotrophic factor in rat visual cortex. Journal of Biological Chemistry. 2003;278(45):44097–44102.
141. Bolton MM, Pittman AJ, Lo DC, Lo Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. Journal of Neuroscience. 2000;20(9):3221–3232.
142. Hewitt SA, Bains JS. Brain-derived neurotrophic factor silences GABA synapses onto hypothalamic neuroendocrine cells through a postsynaptic dynamin-mediated mechanism. Journal of Neurophysiology. 2006;95(4):2193–2198.
143. Yan Z. Regulation of GABAergic inhibition by serotonin signaling in prefrontal cortex: molecular mechanisms and functional implications. Molecular Neurobiology. 2002;26(2-3):203–216.
144. Feng J, Cai X, Zhao J, Yan Z. Serotonin receptors modulate GABAA receptor channels through activation of anchored protein kinase C in prefrontal cortical neurons. Journal of Neuroscience. 2001;21(17):6502–6511.
145. Brandon NJ, Jovanovic JN, Smart TG, Moss SJ. Receptor for activated C kinase-1 facilitates protein kinase C-dependent phosphorylation and functional modulation of GABAA receptors with the activation of G-protein-coupled receptors. Journal of Neuroscience. 2002;22(15):6353–6361.
146. Wang X, Zhong P, Yan Z. Dopamine D4 receptors modulate GABAergic signaling in pyramidal neurons of prefrontal cortex. Journal of Neuroscience. 2002;22(21):9185–9193.
147. Nutt DJ, Cowen PJ, Franklin M. The effect of diazepam on indices of 5-HT function in man. Pharmacology Biochemistry and Behavior. 1986;24(5):1491–1495.
148. Khan A, Haleem DJ. Tolerance in the anxiolytic profile following repeated administration of diazepam but not buspirone is associated with a decrease in the responsiveness of postsynaptic 5-HT-1A receptors. Acta Biologica Hungarica. 2007;58(4):345–357.
149. Hegarty AA, Vogel WH. The effect of acute and chronic diazepam treatment on stress-induced changes in cortical dopamine in the rat. Pharmacology Biochemistry and Behavior. 1995;52(4):771–778.
150. Lambert JJ, Cooper MA, Simmons RDJ, Weir CJ, Belelli D. Neurosteroids: endogenous allosteric modulators of GABAA receptors. Psychoneuroendocrinology. 2009;34(supplement 1):S48–S58.
151. Herd MB, Belelli D, Lambert JJ. Neurosteroid modulation of synaptic and extrasynaptic GABAA receptors. Pharmacology and Therapeutics. 2007;116(1):20–34.
152. Usami N, Yamamoto T, Shintani S, et al. Substrate specificity of human 3(20)α-hydroxysteroid dehydrogenase for neurosteroids and its inhibition by benzodiazepines. Biological and Pharmaceutical Bulletin. 2002;25(4):441–445.
153. Wilson MA, Biscardi R. Effects of gender and gonadectomy on responses to chronic benzodiazepine receptor agonist exposure in rats. European Journal of Pharmacology. 1992;215(1):99–107.
154. Reddy DS, Kulkarni SK. Neurosteroid coadministration prevents development of tolerance and augments recovery from benzodiazepine withdrawal anxiety and hyperactivity in mice. Methods and Findings in Experimental and Clinical Pharmacology. 1997;19(6):395–405.
155. Mirza NR, Nielsen EO. Do subtype-selective gamma-aminobutyric acid A receptor modulators have a reduced propensity to induce physical dependence in mice? Journal of Pharmacology and Experimental Therapeutics. 2006;316(3):1378–1385.
156. Atack JR, Wafford KA, Tye SJ, et al. TPA023 [7-(1, 1-dimethylethyl)-6-(2-ethyl-2H-1, 2, 4-triazol-3-ylmethoxy)-3-(2-fluor ophenyl)-1, 2, 4-triazolo[4, 3-b]pyridazine], an agonist selective for alpha2- and alpha3-containing GABAA receptors, is a nonsedating anxiolytic in rodents and primates. Journal of Pharmacology and Experimental Therapeutics. 2006;316(1):410–422.
157. Knabl J, Witschi R, Hosl K, et al. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature. 2008;451(7176):330–334.
158. Zammit G. Comparative tolerability of newer agents for insomnia. Drug Safety. 2009;32(9):735–748.
159. Ravishankar A, Carnwath T. Zolpidem tolerance and dependence—two case reports. Journal of Psychopharmacology. 1998;12(1):103–104.
160. Sanger DJ, Zivkovic B. Differential development of tolerance to the depressant effects of benzodiazepine and non-benzodiazepine agonists at the omega (BZ) modulatory sites of GABAA receptors. Neuropharmacology. 1992;31(7):693–700.
161. Sanger DJ, Zivkovic B. Investigation of the development of tolerance to the actions of zolpidem and midazolam. Neuropharmacology. 1987;26(10):1513–1518.
162. Perrault G, Morel E, Sanger DJ, Zivkovic B. Lack of tolerance and physical dependence upon repeated treatment with the novel hypnotic zolpidem. Journal of Pharmacology and Experimental Therapeutics. 1992;263(1):298–303.
163. Scherkl R, Kurudi D, Frey HH. Clorazepate in dogs: tolerance to the anticonvulsant effect and signs of physical dependence. Epilepsy Research. 1989;3(2):144–150.
164. Atack JR. Subtype-selective GABAA receptor modulation yields a novel pharmacological profile: the design and development of TPA023. Advances in Pharmacology. 2009;57:137–185.
165. Ator NA. Zaleplon and triazolam: drug discrimination, plasma levels, and self-administration in baboons. Drug and Alcohol Dependence. 2000;61(1):55–68.
(翻訳、訳注:ベンゾジアゼピン情報センター 管理人)
著者:Christiaan Vinkers

Dr. Christiaan Vinkers, MD, PhD
アムステルダム自由大学医療センター精神科、解剖学科、神経科 准教授。GGZ inGeest精神病院の精神科医。主な研究はストレス脆弱性と回復について。
ユトレヒト大学で薬学(2005)、法律(2009)、医学(2009)を学び、 2009年に神経生物学的研究とGABAシステムに関する博士論文を修了。
その後、精神科医になるため臨床研修を開始、精神障害のリスクと回復力、GABAシステムに関する臨床研究を続ける。2014年、ルドルフマグヌス脳センターの精神科医および研究員。2018年よりアムステルダム自由大学医療センター精神科とGGZinGeestt精神病院にて研究員および精神科医。
主な研究目的は、GABAシステムにフォーカスした、エピジェネティック、神経内分泌、脳回路、における対ストレス回復と脆弱性の神経生物学的背景の調査。
外傷性ストレス、特に幼少期におけるそれは、双極性障害、心的外傷後ストレス障害(PTSD)、大うつ病性障害、統合失調症などほとんどの精神障害の危険因子である。さらに、ストレスは脳の構造と機能に持続的な影響を及ぼす。しかし、トラウマ暴露後の結果にはかなりの個人差が見られる。何十年もの研究にもかかわらず、誰が危機的であるか、あるいは回復するかを予測することはできていない。目標はストレス感受性の神経生物学的根拠の追及。
研究のスポンサーには、オランダ科学研究機構(NWO、VENI)、ZonMW、ブレインセンタールドルフマグナス、NARSAD、オランダ脳財団、および神経科学認知ユトレヒトの助成金、がある。
Psychiatry Amsterdamより翻訳